Kystisen fibroosin uudistuva lääkehoito
• Kystinen fibroosi on harvinainen, huonoennusteinen monielinsairaus.
• Sairauden aiheuttava mutaatio CFTR-geenissä johtaa kloridikanavan poikkeavaan toimintaan. Tämä vahingoittaa eniten keuhkoja, mutta myös mm. haimaa, maksaa ja suolistoa.
• Hoito on ollut oireenmukaista, mutta tehokkaimmallakin hoidolla potilaiden elinajan odote on ollut selvästi lyhempi kuin normaaliväestössä.
• Kymmenen viime vuoden aikana markkinoille on tullut mutaatiospesifisiä lääkkeitä, jotka vaikuttavat sairauden perussyyhyn. Niiden ansiosta hoito ja elinajan odote paranevat.


Kystinen fibroosi (CF) on kautta aikain ollut yksi vaikeimmista lasten keuhkosairauksista. Vielä kolmekymmentä vuotta sitten potilaat menehtyivät niin nuorina, että aikuiskeuhkolääkärit eivät heitä juuri ehtineet tavata.
Sairauden aiheuttaa solukalvoilla oleva puutteellisesti toimiva kloridikanavaproteiini (cystic fibrosis transmembrane conductance regulator, CFTR).
Kystinen fibroosi on huonoennusteinen, valkoihoisen väestön yleisin geneettinen sairaus, joka vahingoittaa eniten keuhkoja, mutta vaikuttaa myös haiman, maksan, ruoansulatuskanavan, poskionteloiden, hikirauhasten ja lisääntymiselimien toimintaan (1).
Tautia sairastaa yli 70 000 ihmistä maailmassa, ja ilmaantuvuus on suurin läntisessä Euroopassa ja erityisesti Britanniassa, jossa ilmaantuvuus (1:2 500) on lähes kymmenkertainen Suomen arvioituun ilmaantuvuuteen (1:25 000) verrattuna (2,3). Suomessa potilaita on reilut sata.
Vaikka esiintyvyys vaihtelee suuresti, mikään etninen ryhmä ei ole kokonaan suojassa taudilta. Kystinen fibroosi aiheuttaa paljon sairastavuutta syntymästä lähtien, ja potilaiden elinajan odote on selvästi lyhyempi kuin muun väestön.
Hoito ja elinajan odote ovat pikkuhiljaa tasaisesti kehittyneet parempaan suuntaan, mutta vuonna 2012 otettiin selvä askel eteenpäin, kun oireenmukaisten hoitojen rinnalle saatiin ensimmäiset sairauden perussyyhyn eli kloridikanavan aktiivisuuteen vaikuttavat lääkkeet.
Syksystä 2020 lähtien Euroopassa on myönnetty myyntilupa yhteensä neljälle uudelle CFTR-proteiinin synteesiin tai toimintaan vaikuttavalle lääkkeelle eli CFTR:n muuntajalle. Tutkimusputkessa on erilaisia kohteita lääkeainemolekyyleistä geeniterapiaan, mutta tässä kirjoituksessa keskitytään nyt myyntiluvan saaneisiin lääkkeisiin, joille Lääkkeiden hintalautakunta myönsi 4.2.2021 rajoitetun peruskorvattavuuden.
Patogeneesi
Kystinen fibroosi johtuu CFTR-geenin mutaatiosta, joka vaikuttaa CFTR-proteiinin synteesiin. Sairastunut lapsi on perinyt kaksi viallista CFTR-geeniä, yhden kummaltakin vanhemmalta (4). Sairautta aiheuttavia mutaatioita tunnetaan eri lähteiden mukaan noin 2 000. Mutaatio voi aiheuttaa CFTR-kanavan aktiivisuuden vähenemistä useilla mekanismeilla.
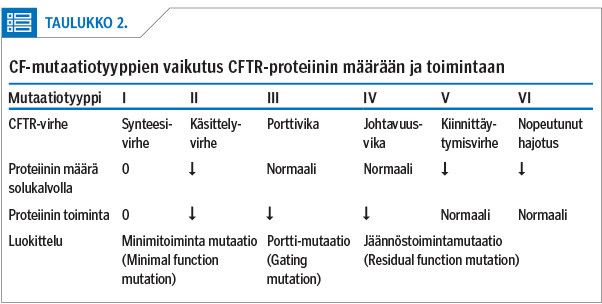
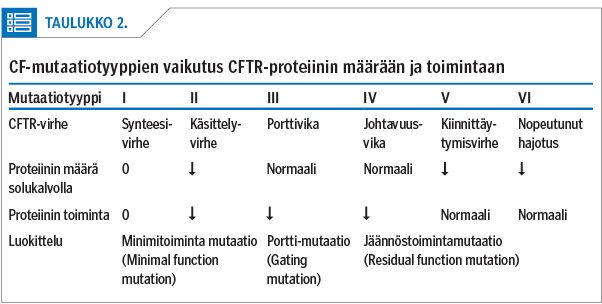
Mutaatiot jaetaan perinteisesti kuuteen eri tyyppiin (5,6) sen perusteella, millaisen häiriön ne proteiinisynteesissä aiheuttavat (taulukko 1). Geenimutaation tyyppi on tärkein taudin kliiniseen kuvaan ja vaikeusasteeseen vaikuttava tekijä, mutta myös muilla geneettisillä ominaisuuksilla sekä ympäristötekijöillä on merkitystä.
Geenimutaation aiheuttama proteiinisynteesin häiriö saa epiteelisolujen apikaalipinnoilla olevien kloridikanavien aktiivisuuden vähenemään. Tällöin kanavan läpi kulkevien kloridi- ja bikarbonaatti-ionien määrä vähenee, mikä johtaa solun tuottamien nesteiden poikkeavaan eritykseen.
Soluja, joissa nesteiden eritys on mutaation vuoksi poikkeava, on mm. hengitysteissä, iholla, ruoansulatuselimistössä ja lisääntymiselimissä. Keuhkoissa tämä tarkoittaa ilmateiden juoksevan pintanesteen muuttumista paksuksi, sitkeäksi, tukkivaksi limaksi ja haimassa ruoansulatusentsyymien vähentynyttä erittymistä suoleen. Kyseessä on siis monielinsairaus, jossa vakavimmat ongelmat ovat etenevä keuhkovaurio sekä haiman vajaatoimintaan liittyvä ravintoaineiden ja vitamiinien imeytymishäiriö (1).
Kliininen kuva
Kystisen fibroosin aiheuttama keuhkosairaus alkaa jo vastasyntyneenä. Tämä osoitettiin jo 1940-luvulla suolentukkeumaan kuolleiden vastasyntyneiden ruumiinavauksissa (7). Myöhemmin keuhkovaurioihin liittyvät muutokset imeväisillä ja pikkulapsilla on pystytty osoittamaan keuhkojen kuvantamistutkimuksissa, keuhkofunktiokokeissa ja bronkoalveolaarisessa huuhtelunesteessä (8).
CFTR-kanavan puutteellisen toiminnan vuoksi syntyvä sitkeä lima tukkii keuhkoissa pieniä ilmateitä, mikä nähdään ilmasalpauksena. Lisäksi lima heikentää värekarvojen toimintaa. Poikkeava CFTR-kanavan toiminta johtaa myös asidoosiin, jota pidetään yhtenä syynä heikentyneeseen immuunipuolustukseen keuhkoissa. Bakteerikolonisaatiot ja -infektiot ylläpitävät neutroliifista inflammaatiota, joka johtaa aluksi ilmateiden mikroskooppisiin muutoksiin ja sittemmin makroskooppisiin keuhkoputkien laajentumiin eli bronkiektasioihin (9).
Sairaus ilmenee eri iässä eri tavalla. Vastasyntyneellä kystinen fibroosi voi ilmetä leikkausta vaativana suolitukoksena, ja noin 12–25 % sitä sairastavista vauvoista päätyy kiireelliseen leikkaukseen jo heti synnyttyään (10). Ensimmäisten elinviikkojen aikana sairaus usein oireilee siten, ettei vauvan paino nouse, mikä johtuu haiman vajaatoiminnan aiheuttamasta ravintoaineiden huonosta imeytymisestä. Lisäksi vauvalla saattaa olla vaikeita elektrolyyttihäiriöitä, jotka voivat johtaa yleistilan voimakkaaseen heikentymiseen ja jopa tehohoitoon.
Hengitystieoireet, kuten jatkuvat infektiot, hengitysteiden limaisuus, yskä ja hengenahdistus, alkavat usein jo varhaislapsuudessa. Viimeistään teini-iässä keuhkoissa nähdään eriasteisia bronkiektasioita. Keuhkojen bakteeri-infektiot hankaloituvat keuhkojen tilanteen heikentyessä ja yhä vastustuskykyisemmät bakteerit kolonisoivat ja infektoivat keuhkoja.
Keuhko-ongelmien lisäksi potilailla on usein haiman vajaatoiminta. Osalle kehittyy muita liitännäissairauksia, kuten maksan fibrotisoituminen sappiteiden tukkeutumisen vuoksi, diabetes haimateiden tukkeutumisen ja insuliinierityksen huononemisen vuoksi, nenän polypoosi tai osteoporoosi D-vitamiinin imeytymishäiriön vuoksi (9).
Oireenmukainen hoito
Kystisen fibroosin perinteinen hoito perustuu lähinnä oireiden hoitamiseen, ravitsemustilan ylläpitämiseen ja keuhkovaurioiden ehkäisemiseen. Keuhkojen hoito perustuu ilmateiden liman poistamiseen inhaloimalla hypertonista keittosuolaliuosta (11) ja dornaasi-alfaa (12). Liman irtoamista voidaan edistää myös fysioterapialla ja rasittavalla liikunnalla. Pahenemisvaiheet ja bakteeri-infektiot on hoidettava antibiooteilla. Lisäksi potilailla on käytössä haimaentsyymit (Creon), A-, E-, K ja D-vitamiinilisät, ravintolisät, maksasairauden ja refluksitaudin lääkitykset sekä tarvittaessa insuliini ja osteoporoosin hoitolääkkeet (13).
Päivittäinen hoito ja hyvän ravitsemustilan ylläpito vaatii perheeltä tiukkaa rutiinia ja on kuormittavaa. Lasta tai nuorta voi olla vaikea motivoida päivittäisiin limanpoistorutiineihin.
Tehokkaimmasta mahdollisesta perinteisestä hoidosta huolimatta keuhkovaurio etenee lähes poikkeuksetta. Jotkin hankalat moniresistentit bakteerit, kuten pseudomonas, Stenotrophomonas maltophilia ja ei-tuberkuloottiset mykobakteerit, nopeuttavat etenemistä entisestään. Keuhkoinfektioihin kohdistetut antibioottihoidot ovat pitkiä ja lisäävät hoidon rasitusta. Keuhkonsiirto on mahdollinen joillekin potilaille keuhkotoiminnan heikennyttyä.
Usein kotona aletaan sitoutua kunnolla hoitoon vasta keuhkotoiminnan huononuttua merkittävästi. Toistuvat suonensisäiset antibioottihoidot, painonlasku, PEG-letkun asettaminen ja insuliinihoidon aloitus kasaantuvat jo valmiiksi väsyneelle potilaalle ja kuormittuneelle perheelle. Kaiken kaikkiaan edenneen kystisen fibroosin hoito on erittäin vaativaa ja potilasta psyykkisesti kuormittavaa. Hoitoväsymys on keskeinen perinteisen hoidon ongelma.
Uudet lääkkeet
Pian CFTR-geenin löytymisen jälkeen havaittiin, että CFTR-proteiinin toimintaa voi stimuloida erilaisilla molekyyleillä, esimerkiksi joillain flavonoideilla. Tutkimuksessa genistiini kykeni lisäämään mutatoituneen Gly551ASP-CFTR-kanavan aukioloa in vitro yli kymmenkertaiseksi niin, että se vastasi normaalin kanavan aktiivisuutta (14). Tämä oli tärkeä osoitus siitä, että kanavan aktiivisuuteen pystytään vaikuttamaan, ja se loi pohjan tulevalle lääkekehitykselle (9).
Maailman yleisin CFTR-mutaatio on F508del, joka johtaa fenyylialaniinin puuttumiseen kohdassa 508. Proteiinisynteesissä viallinen proteiini tunnistetaan endoplasmisessa retikulumissa jo ennen kuin proteiinin luenta on valmis ja proteiinin alku tuhotaan proteosomeissa. Pieni määrä proteiinia ohittaa tämän ensimmäinen laatukontrollin ja päätyy solukalvolle, josta se kuitenkin tunnistetaan virheelliseksi, poistetaan ja tuhotaan lysosomeissa (9).
Vuonna 1996 kyettiin ensimmäistä kertaa osoittamaan in vitro, että osa mutatoituneesta F508del-CFTR:sta pystyttiin pelastamaan solun laatukontrollin laukaisemalta tuhoamiselta alhaisella lämpötilalla ja glyserolilla (15). Parikymmentä vuotta ja satojatuhansia molekyylejä myöhemmin ensimmäinen "korjaaja" eteni kliinisiin tutkimuksiin 2015 (9).
Kystisen fibroosin uusia lääkkeitä kutsutaan yhteisnimellä CFTR:n muuntajat. Kanavaproteiinin vähentynyt aktiivisuus voi johtua joko kanavaproteiinin vähentyneestä määrästä solukalvolla tai sen vähentyneestä toiminnasta (16,18) (taulukko 2). CFTR:n muuntajat ovat joko korjaajia, jotka pyrkivät korjaamaan proteiinin synteesiä ja siten saamaan solukalvolle mahdollisimman paljon toimivaa CFTR-proteiinia, tai potentiaattoreita, jotka pyrkivät tehostamaan virheellisen kanavan johtavuutta (17).
Ivakaftori
Ensimmäinen CFTR-kanavan aktiivisuutta tehostava potentiaattori ivakaftori (Kalydeco; Vertex Pharmaceuticals) sai FDA:n (US Food and Drug Administration) ja Eman (European Medicine Agency) myyntiluvat vuonna 2012. Molekyyli oli osoittanut tehonsa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa potilailla, joilla oli tyypin III porttimutaatio Gly551ASP-CFTR (19).
Tutkimuksessa todettiin nopea ja kestävä keuhkojen toiminnan paraneminen: keuhkofunktiotutkimuksessa nopea sekuntikapasiteetti (FEV1) parani 10 % ja tulos säilyi tutkimuksen loppuun saakka. Lääkeryhmässä olleiden hien kloridipitoisuus pieneni normaalitasolle (keskimääräinen alenema 48 mmol/l lähtötasosta), ja 48 viikon seurannassa potilailla oli 55 % vähemmän pahenemisvaiheita. Lisäksi painonkehitys oli parempaa (nousu 2,7 kg) ja elämänlaatu parani oirekyselyssä (CFQ-R) (19). Haittavaikutuksissa ei ollut eroja lume- ja lääkeryhmän välillä.
Myöhemmin ivakaftorin teho on osoitettu muissakin porttimutaatioissa ja tutkimukset ovat käynnissä myös jäännösmutaatioissa. Koska lääkkeen on osoitettu tehostavan myös muiden kuin porttimutaation vuoksi vaurioituneiden CFTR-kanavien toimintaa, se on osana mukana kaikissa CFTR:n muuntajissa.
Ivakaftorilla on Euroopassa myyntilupa monoterapiana homo- tai heterotsygoottisessa porttimutaatiossa 4 kuukauden iästä lähtien ja yhdistelmänä muiden CFTR:n muuntajien kanssa. Monoterapiasta hyötyy noin 5 % potilaista, mutta Suomessa ei ole toistaiseksi tiedossa potilaita, joilla olisi tämä mutaatio.
Lumakaftori-ivakaftori
Ensimmäinen ns. korjaajan ja potentiaattorin yhdistelmä, lumakaftori-ivakaftori (Orkambi, Vertex Pharmaceuticals), kehitettiin maailman yleisimmän mutaation, homotsygoottisen F508del-geenivirheen aiheuttaman kloridikanavan toiminnanvajauksen korjaamiseksi. Se sai myyntiluvat vuonna 2015.
Satunnaistetussa, lumekontrolloidussa kaksoissokkotutkimuksessa sen todettiin pienentävän hien kloridipitoisuutta jonkin verran, keuhkofunktiotutkimuksissa FEV1 parani 2,6–4 % ja pahenemisvaiheet vähenivät 30–39 % seurannassa, joka kesti 48 viikkoa. Haittavaikutusten ilmaantuvuudessa ei todettu merkittävää eroa lume- ja lääkeryhmän välillä, mutta joitain infektiivisiä pahenemisvaiheita, kohonnutta verenpainetta ja maksa-arvojen nousua raportoitiin (20).
Tulokset porttimutaatiossa olivat kuitenkin heikommat kuin ivakaftorin, ja siksi lumakaftori-ivakaftorin käyttöönotto useissa maissa (mm. Britanniassa) viivästyi, koska hyötyä suhteessa kalliiseen hintaan pidettiin vähäisenä. Tällä hetkellä lumakaftori-ivakaftorilla on myyntilupa yli 2-vuotiaille potilaille, joilla on homotsygoottinen F508del-mutaatio.
Tetsakaftori-ivakaftori
Seuraavassa, vuonna 2017 myyntiluvan saaneessa lääkkeessä lumakaftorin tilalle oli vaihdettu tetsakaftori (Symkevi, Vertex Pharmaceuticals). Lääkkeellä osoitettiin olevan tehoa sekä homotsygoottisessa että heterotsygoottisessa F508del-mutaatiossa, jossa toisena mutaationa on ns. jäännösmutaatio.
Kahdessa eri tutkimuksessa tetsakaftori-ivakaftorin osoitettiin parantavan jonkin verran keuhkofunktiota (FEV1 parani 4 %) ja vähentävän pahenemisvaiheita 35 % (21,22). Tetsakaftorin tehossa ei todettu merkittävää eroa lumakaftoriin verrattuna, mutta haittavaikutuksia todettiin vähemmän. Esimerkiksi kohonnutta verenpainetta ei havaittua tetsakaftoria saavilla potilaille. Myös lääkkeiden yhteisvaikutuksia tetsakaftorilla on vähemmän kuin lumakaftorilla.
Tetsakaftori-ivakaftorilla on myyntilupa Euroopassa yli 6-vuotiaille joko homotsygoottisissa tai heterotsygoottisissa F508del-mutaatioissa, joilla on toisena mutaationa joku 14 luetellusta jäännösmutaatiosta (23).
Eleksakaftori-tetsakaftori-ivakaftori
Vuonna 2019 julkaistiin ensimmäiset tulokset kolmen molekyylin yhdistelmästä, eleksakaftori-tetsakaftori-ivakaftorista (Kaftrio, Vertex Pharmaceuticals). Kolmanneksi molekyyliksi lisätty eleksakaftori on tetsakaftorin tapaan korjaaja. Toisin kuin tetsakaftori, joka kykenee korjaamaan vain F508del-CFTR-proteiinin synteesiä, eleksakaftori kykenee lisäämään CFTR-kanavaproteiinin määrää solukalvolla myös potilailla, joilla on minimitoimintamutaatio.
Kolmoishoito todettiin tehokkaaksi potilaille, joilla on F508del-mutaatio homotsygoottisena (24). Näillä potilailla FEV1-arvo parani 10,4 %, kun se lumeryhmässä parani vain 0,4 %. Yhdistelmä osoittautui tehokkaaksi myös heterotsygoottisessa muodossa, jossa toinen mutaatio on ns. minimitoimintamutaatio (minimal function mutation) (25). Tutkimukseen osallistui yhteensä 403 potilasta. 24 hoitoviikon jälkeen kolmoishoitoa saaneilla FEV1 parani 13,9 % (lumeryhmässä 0,4 %). Lisäksi hien kloridi-ionipitoisuus pieneni (39–44 mmol/l), ravitsemustila koheni (BMI nousi 1,13 kg/m2) ja elämänlaatu parani.
Maailmanlaajuisesti jopa 90 % kystistä fibroosia sairastavista voi hyötyä kolmoishoidosta (26). Hyvät tulokset ja kolmoishoidon laaja käytettävyys ovat vauhdittaneet sen käyttöönottoa useissa länsimaissa. Tällä hetkellä eleksakaftori-tetsakaftori-ivakaftorilla on myyntilupa ainoastaan yli 12-vuotiaille potilaille. FDA ja nyt myös Ema ovat hyväksyneet käyttöaiheiksi kaikki heterotsygoottiset F508del-mutaatiot riippumatta toisesta mutaatiosta.
Käyttö ja varotoimet
Käyttöaiheiden tuntemisen lisäksi CFTR:n muuntajien käyttöönotto vaatii erityisosaamista. Lääkkeen aloituksen yhteydessä suositellaan tutkittavaksi perusverikokeet sekä maksa- ja munuaiskokeet. Alle 18-vuotiaille suositellaan silmälääkärin tarkistusta ennen tetsakaftorihoidon alkua ja hoidon aikana vuosittain. Hoitovasteen arvioimiseen suositellaan hikikoetta ja keuhkofunktiotutkimuksia.
Orkambi annetaan 2–5-vuotiaille rakeina, 1 raepussiannos 100 mg lumakaftoria ja 125 mg ivakaftoria otetaan 12 tunnin välein. Orkambi-tabletit sisältävät lumakaftoria 100 mg tai 200 mg ja ivakaftoria 125 mg. Annostus 6–11-vuotiaille on 100 mg/125 mg 2 tablettia 12 tunnin välein ja 12 vuodesta lähtien 200 mg/125 mg 2 tablettia 12 tunnin välein.
Yksi Symkevi-tabletti sisältää 100 mg tetsakaftoria ja 150 mg ivakaftoria. Suositeltu vuorokausiannos on yksi tabletti aamulla ja yksi 150 mg:n ivakaftoriatabletti Kalydeco illalla (12 tunnin kuluttua).
Yksi Kaftrio-tabletti sisältää 100 mg eleksakaftoria, 50 mg tetsakaftoria ja 75 mg ivakaftoria. Se otetaan niin ikään Kalydecon (150 mg) kanssa. Suositeltu vuorokausiannos on kaksi Kaftrio-tablettia aamulla ja yksi Kalydeco 150 mg illalla (noin 12 tunnin kuluttua). Kaikki CFTR:n muuntajat suositellaan otettavaksi rasvaisen ruoan kanssa.
Hoidon aikana tulee seurata verikokeita ja erityisesti maksa-arvoja. Vähäistä maksa-arvojen nousua on kuvattu yleisenä haittavaikutuksena, mutta myös yksittäisiä hankalampia lääkeainehepatiitteja on esiintynyt. CFTR:n muuntajat metaboloituvat mm. CYP3A4-entsyymin kautta, joten niillä on lukuisia interaktiota muiden lääkkeiden, kuten joidenkin antibioottien ja sienilääkkeiden kanssa. Tämän vuoksi muiden lääkkeiden määräämisessä tarvitaan erityistä tarkkuutta.
Korvattavuus
Suomessa lääkkeiden hintalautakunta myönsi 4.2.2021 rajoitetun peruskorvattavuuden kaikille neljälle CFTR:n muuntajavalmisteelle. Potilaille, joilla on F508del-mutaatio homotsygoottisena, korvattavia ovat Orkambi (2–11-vuotiaille) sekä Symkevi ja Kaftrio yli 12-vuotiaille. Kaftrio korvataan myös yli 12-vuotiaille potilaille, joilla on F508del-mutaatio heterotsygoottisena yhdessä minimitoimintamutaation kanssa. Ivakaftori korvataan yhdistelmähoidossa, mutta ei monoterapiana. Odotamme korvattavuuden laajenemista kaikille potilaille, joilla on F508-mutaatio heterotsygoottisena.
Lopuksi
Pitkäaikaistuloksia uusista lääkkeistä ei ole vielä ehtinyt juuri tulla. Joitain julkaisuja ivakaftorilääkityksen 5 vuoden seurannasta on, ja niiden valossa lääke on ollut hyvin siedetty, merkittäviä haittavaikutuksia ei ole ollut ja keuhkofunktio näyttäisi säilyvän ennallaan seurannan aikana (27,28). Lisäksi uusien lääkkeiden on osoitettu vähentävän infektioiden aiheuttamia pahenemisvaiheita, parantavan ravitsemustilaa, haiman toimintaa ja elämänlaatua (27,28).
Seurannan tuloksia voidaan joutua odottamaan pidempään. Vastasyntyneenä potilaan keuhkot ovat normaalit, mutta kystinen fibroosi alkaa jo varhain ja johtaa ajan myötä hengitysteiden arpeutumiin ja bronkiektasioihin. Näitä muutoksia pidetään peruuttamattomina, joten ehkäisy voi olla tehokkaampaa kuin edenneen sairauden hoito. Kaikkein tehokkain kolmoishoito on nyt tarjolla yli 12-vuotiaille, ja tutkimukset nuoremmista potilaista ovat käynnissä.
Kaikkia potilaita CFTR:n muuntajat eivät voi auttaa. Maailmanlaajuisesti noin 10 % potilaista on lääkehoidon ulottumattomissa laajimminkin käyttöaihein, ja Suomessa osuus on noin 30 %.
Edenneen kystisen fibroosin hoito on erittäin vaativaa ja potilasta psyykkisesti kuormittavaa. Suomessa hoitoa on viime aikoina pyritty keskittämään yliopistosairaaloihin, koska se vaatii moniammatillista erityisosaamista. Uusien lääkkeiden ansiosta sairauden hoito ja elinajan odote paranevat.
Kirsi Malmivaara: Apuraha (Hengityssairauksien tutkimussäätiö), luentopalkkiot (CF-yhdistys, Filha, Vertex), matka-, majoitus- ja kokouskulut (Vertex).
Varpu Elenius: Luentopalkkiot (CF-yhdistys, Novartis, Sanofi, Thermo Fisher, Vertex).
- 1
- O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009;373:1891–904.
- 2
- Cystic Fibrosis Foundation; Bethesda, USA. 5.5.2021: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/
- 3
- Halme M, Kajosaari M. Kystinen fibroosi – harvinainen monielinsairaus. Duodecim 2006;122:1341–6.
- 4
- Rommens JM, Iannuzzi MC, Kerem B ym. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059–65.
- 5
- Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993;73:1251–4.
- 6
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005;352:1992–2001.
- 7
- Zuelzer WW, Newton WA. The pathogenesis of fibrocystic disease of the pancreas; a study of 36 cases with special reference to the pulmonary lesions. Pediatrics 1949;4:53–69.
- 8
- Belessis Y, Dixon B, Hawkins G ym. Early cystic fibrosis lung disease detected by bronchoalveolar lavage and lung clearance index. Am J Respir Crit Care Med 2012;185:862–73.
- 9
- Mall M, Elborn JS. ERS monograph, Cystic Fibrosis. Norwick UK, European respiratory society 2014:1–13.
- 10
- Meghana S, Roderick H. Meconium ileus in cystic fibrosis. Review J Cyst Fibros 2017;16 suppl 2:S32–S39.
- 11
- Elkins MR ym. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354:229–40.
- 12
- Yang C1, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2018 Sep 6.
- 13
- Castellani C ym. ECFS best practice guidelines: the 2018 revision. J Cystic Fibrosis 2018;17:153–78.
- 14
- Illek B, Zhang L, Lewis NC, Moss RB, Dong JY, Fischer H. Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am J Physiol 1999;277:C833–9.
- 15
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 1996;271:635–8.
- 16
- Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013;1:158–63.
- 17
- Cuevas-Ocaña S, Laselva O, Avolio J, Nenna R. The era of CFTR modulators: improvements made and remaining challenges. Breathe (Sheff) 2020;16(2):200016. doi: 10.1183/20734735.0016-2020
- 18
- Rowe SM, Daines C, Ringhausen FC ym. Tezacaftor-ivacaftor in patients with cystic fibrosis and Phe508del and a residual function mutation. N Engl J Med 2017;377:2024–35.
- 19
- Ramsey BW, Davies J, McElvaney NG ym. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–72.
- 20
- Wainwright CE, Elborn JS, Ramsey BW ym. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015;373:220–31.
- 21
- Taylor-Cousar JL ym. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017;377:2013–23.
- 22
- Rowe SM ym. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 2017;377:2024–35.
- 23
- European Medicines Agency, Amsterdam, Hollanti, 22.1.2021.https://www.ema.europa.eu/en/medicines/human/EPAR/symkevi
- 24
- Heijerman H, McKone E, Downey D ym. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394:1940–8.
- 25
- Middleton P, Mall M, Dřevínek P ym. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019;381:1809–19.
- 26
- Southern KW, Murphy J, Sinha IP, Nevitt SJ. CFTR correctors, a therapy for cystic fibrosis targeted at specific variants (most commonly F508del). Cochrane Database Syst Rev; 12: CD010966, 17/12/2020.
- 27
- Higgins M, Volkova N, Moy K, Marshall B, Bilton D. Real-world outcomes among patients with cystic fibrosis treated with ivacaftor: 2012-2016 experience. Pulm Ther 2020;6:141–9.
- 28
- Dave K, Dobra R, Scott S ym. Entering the era of highly effective modulator therapies. Pediatr Pulmonol 2021;56 suppl 1:S79–S89.
Advances in drug therapy for cystic fibrosis
Cystic fibrosis (CF) is a rare and life-shortening multi-organ disease with poor prognosis. The disease is caused by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, resulting in abnormal function of the chloride channel. The most severe damage is caused to the lungs, alongside serious damage to the pancreas, liver, kidneys and intestines. Typically, treatment has been symptomatic but even with the most effective treatment, life expectancy in CF patients has been notably shorter than in the general population. The new mutation-specific orally taken drugs target the CFTR by enhancing its function. With these new CFTR modulators, now available in Finland, management of the disease and life expectancy will be improved.







{kind=link}
{kind=link}