Lasten munuaissairaudet
Lasten munuaissairaudet voivat olla synnynnäisiä kehityshäiriöitä, munuaiskerässairauksia tai tubulaarisia sairauksia.
Ne ovat merkittävä erotusdiagnostinen vaihtoehto selvitettäessä lapsen kasvuhäiriön, huonon menestymisen tai korkean verenpaineen syytä.
Lasten vaikeiden munuaiskorvaushoitoon johtavien sairauksien ilmaantuvuus on pysynyt vakaana, mutta munuaisten vajaatoiminnan syyt ovat osittain muuttuneet.
Vaikeaakin munuaissairautta sairastavien lasten eloonjäämisennuste on hyvä ja heitä siirtyy yhä enemmän aikuisten terveydenhuollon seurantaan.

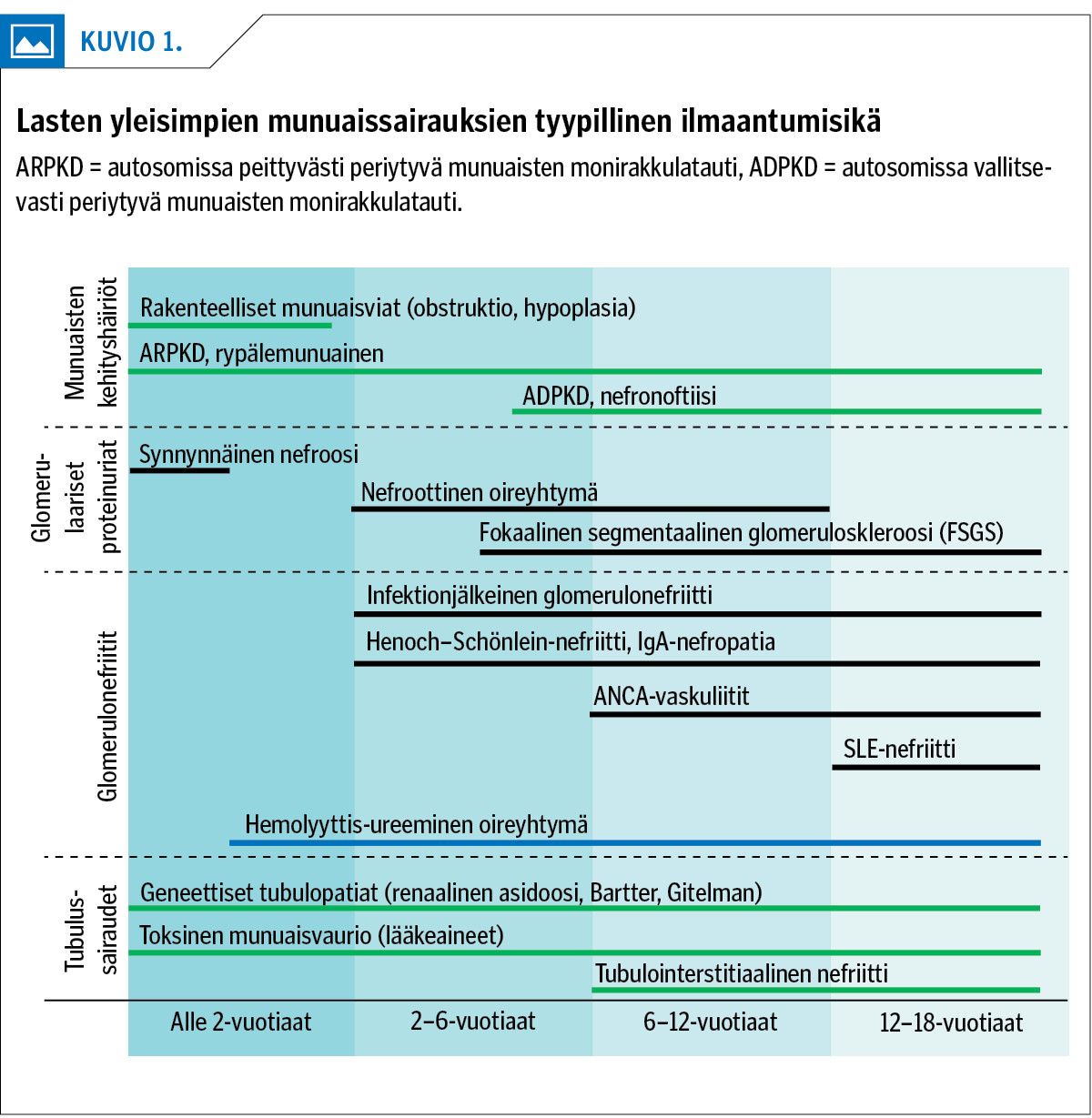

Lapsilla munuais- ja virtsateiden sairaudet ovat melko yleisiä. Pelkästään virtsatietulehdukseen sairastuu vuosittain satoja lapsia. Sen sijaan lasten vakavat, munuaisten vajaatoimintaan johtavat sairaudet ovat harvinaisia. Diagnostisten menetelmien ja hoitokeinojen kehittyminen on parantanut lasten munuaissairauksien ennustetta. Tästä huolimatta keinomunuaishoitoa ja munuaisensiirtoa tarvitsevia lapsia ja nuoria on Suomessa vuosittain 15–20 (1).
Lasten munuaissairauksien diagnostiikka, hoito ja seuranta on lastenlääkärien, lastennefrologien ja lastenurologien yhteistyötä. Munuaissairauksien varhainen toteaminen on ennusteen kannalta tärkeää, ja siinä perusterveydenhuollon rooli on merkittävä (taulukko 1). Aikuistuvien munuaissairaiden nuorten määrän lisääntyessä jatkoseurannan tarpeen arviointi ja seurannan järjestäminen ovat kasvava haaste.
Munuaissairauksien tyypit ja oireisto
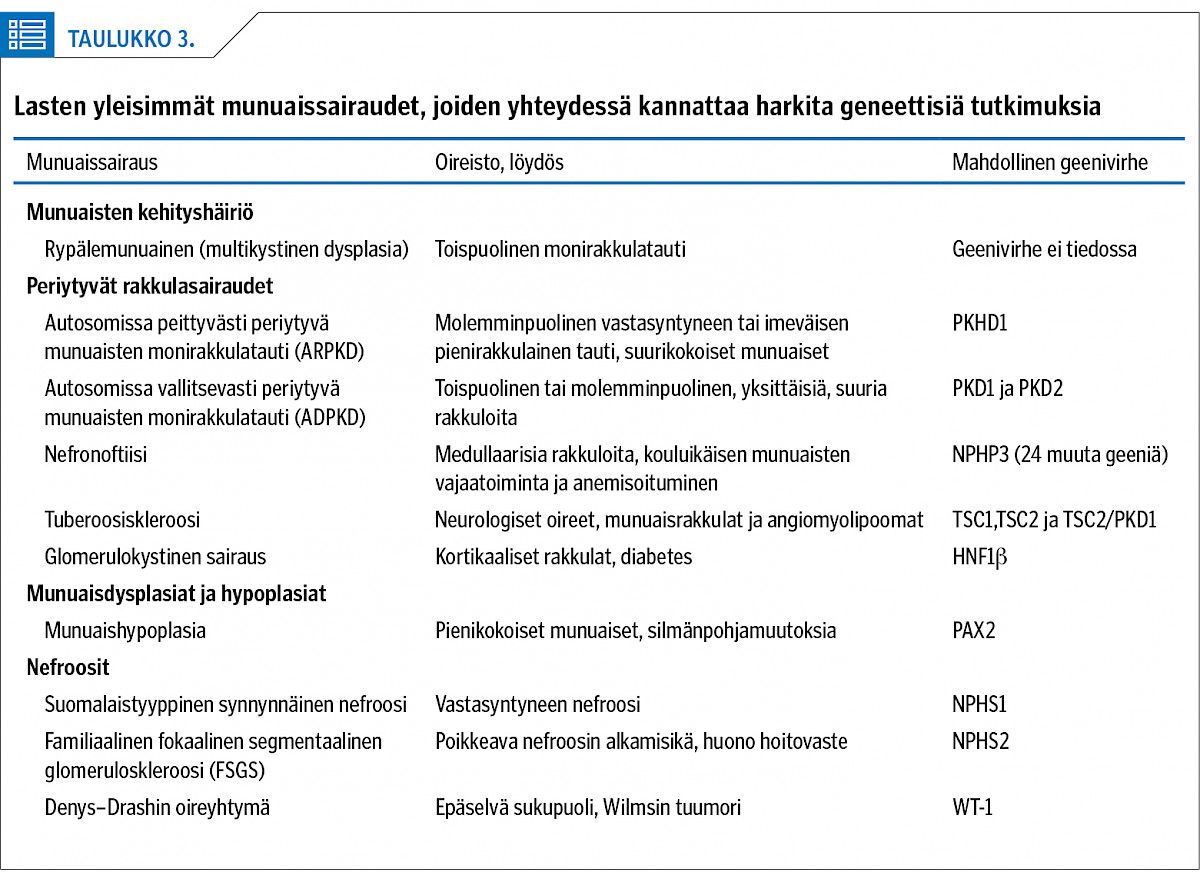
Lasten munuaissairaudet voidaan jakaa syntymekanismin perusteella synnynnäisiin munuaisten ja virtsateiden kehityshäiriöhin, munuaiskerässairauksiin ja tubulussairauksiin (2). Osa munuaissairauksista on perinnöllisiä, ja uudet genetiikan tutkimusmenetelmät ovat tarkentaneet niiden diagnostiikkaa.
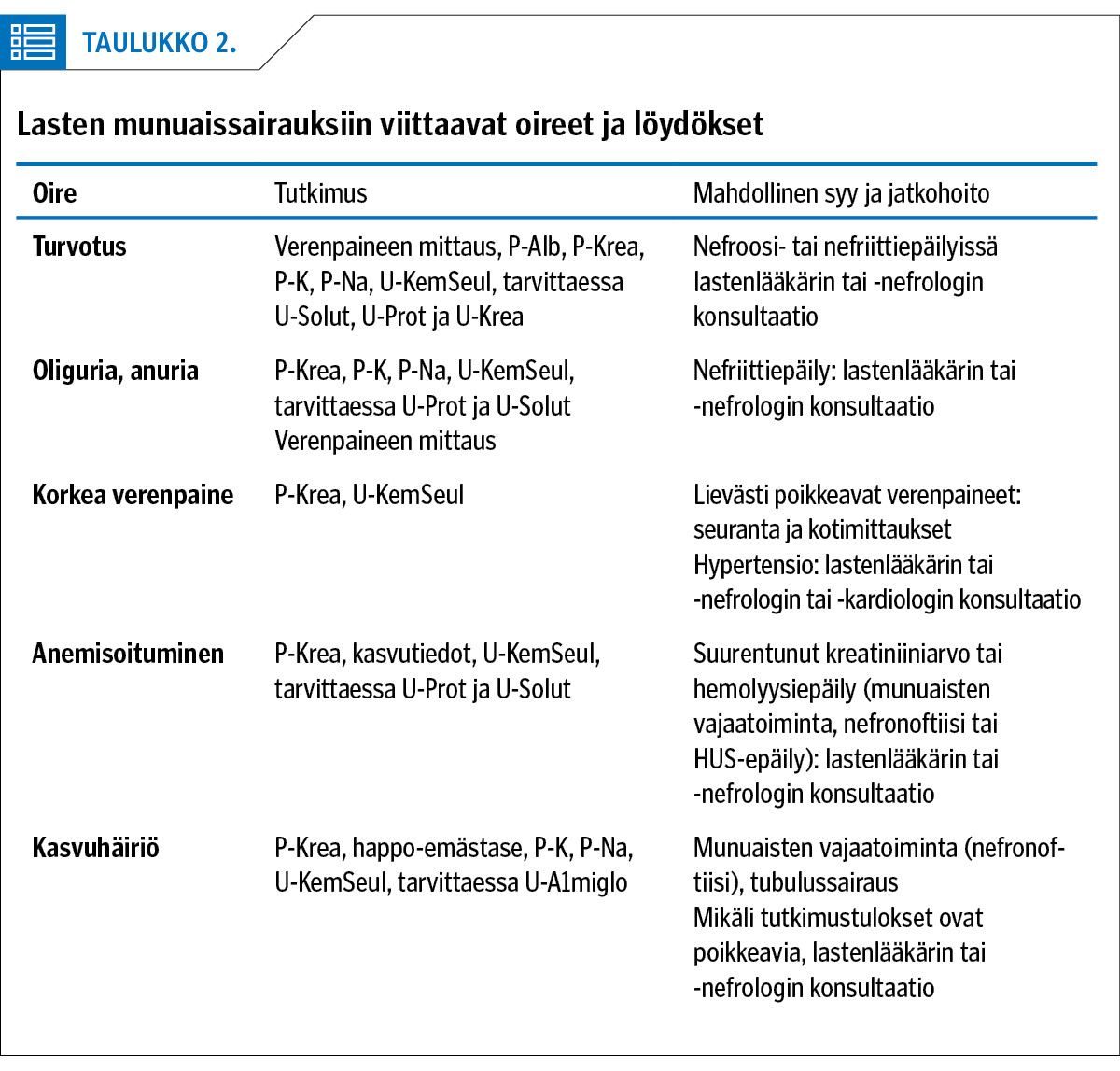
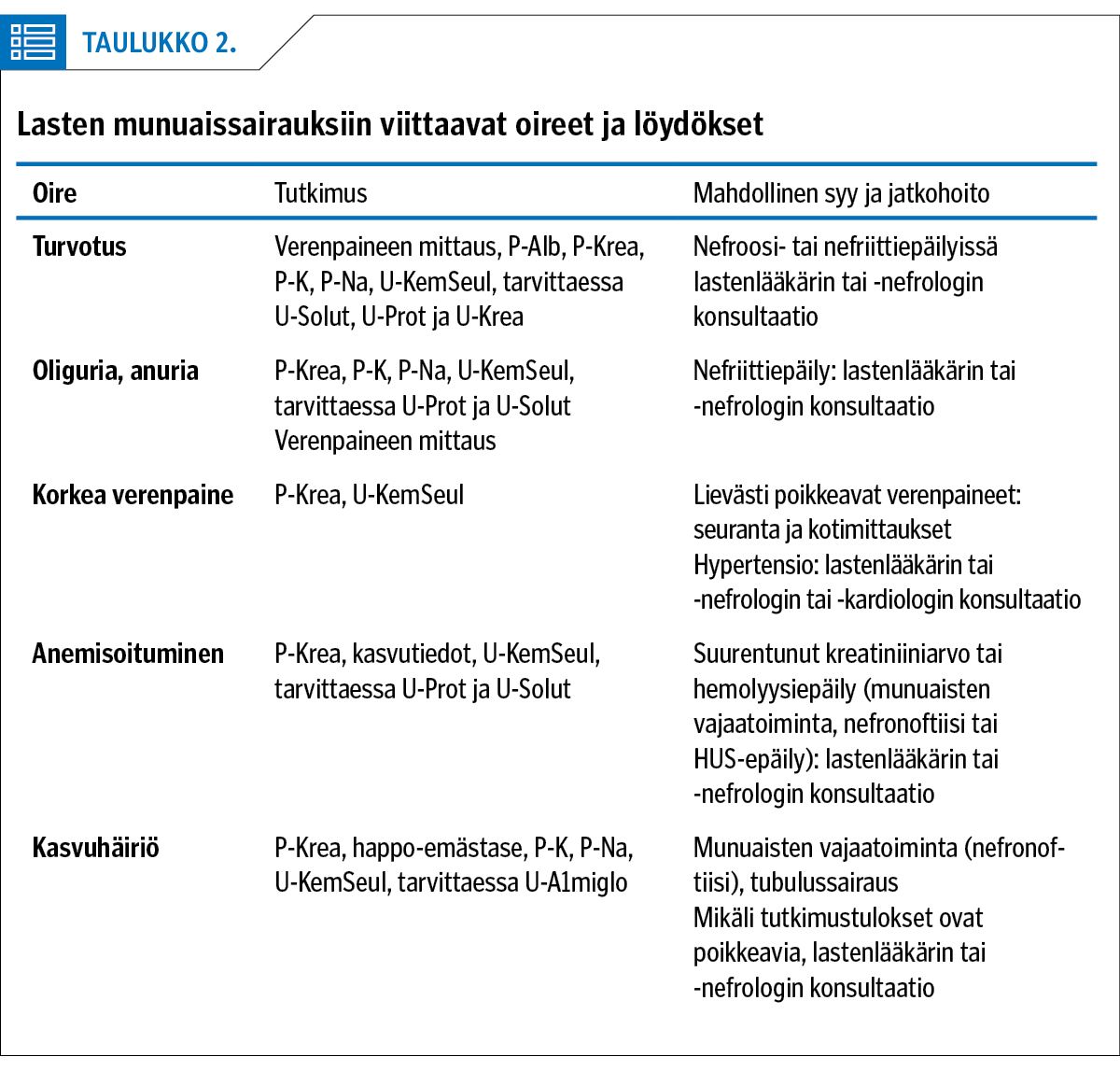
Munuaissairauden ensioireet ja löydökset ovat hyvin vaihtelevia (taulukko 2). Munuaiskerästen sairauksiin viittaavat nefriittioireet eli veri- ja valkuaisvirtsaisuus, turvotukset ja vähentynyt virtsaneritys. Tubulaariset sairaudet ilmenevät usein lisääntyneenä virtsanerityksenä ja sen seurauksena runsaana juomisena. Niihin liittyy myös monesti laihtumista ja huonoa menestymistä ikätovereihin verrattuna. Munuaissairaudet onkin syytä pitää mielessä pohdittaessa syitä edellä mainittuihin oireisiin ja löydöksiin.
Munuaissairaus voidaan todeta sattumalöydöksenä seulontatutkimusten tai muun oireen selvittelyjen yhteydessä (2,3). Virtsan poikkeava ulkonäkö (punainen tai rusehtava väri, runsas vaahtoaminen) voi viitata munuaisten tai virtsateiden sairauteen. Nämä löydökset on helppo varmentaa virtsan kemiallisella seulonnalla (liuskatesti) ja laajentaa tutkimuksia tarvittaessa (3).
Munuaisten ja virtsateiden rakenteelliset viat
Virtsaelinten synnynnäiset kehityshäiriöt todetaan usein raskaudenaikaisissa sikiön rakenneultraäänitutkimuksissa (4), ja vain noin 15 %:lla poikkeavuus todetaan syntymän jälkeen esimerkiksi virtsatietulehduksen yhteydessä (kuvio 1). Kehittyvissä maissa syntyneillä Suomeen saapuneilla lapsilla virtsaelinten kehityshäiriöt on syytä pitää mielessä erotusdiagnostisena vaihtoehtona pohdittaessa munuaisen vajaatoiminnan syytä.
Virtsaelinten kehityshäiriöitä voi esiintyä sekä munuaisissa että virtsateissä tai ne voivat rajoittua jompaankumpaan. Osaan voi liittyä poikkeavuuksia myös muissa elinjärjestelmissä. Virtsaelinten kehityshäiriöön johtavia geenivirheitä on toistaiseksi tiedossa vain muutamia, ja ne ovat transskriptiotekijöiden, kuten PAX2, HNF1β ja SALL1, mutaatioita (5).
Munuaisten kehityshäiriöt ilmenevät tyypillisesti toisen tai kummakin munuaisen kudosrakenteen häiriintymisenä (dysplasia), pienikokoisuutena (hypoplasia) tai munuaisen puuttumisena (agenesia). Virtsateiden poikkeavuudet ovat yleisimmin virtsateiden ahtaumia tai laajentumia. Niihin voi liittyä sukupuolielinten ja mahdollisesti suoliston poikkeavuuksia. Virtsan takaisinvirtaus (vesikoureteraalinen refluksi, VUR) voi esiintyä itsenäisenä löydöksenä tai osana laajempaa virtsaelinten kehityshäiriötä.
Virtsan takaisinvirtauksen merkitystä munuaisvaurion aiheuttajana on arvioitu useissa tutkimuksissa (6). Todennäköisimmin erityisesti pojilla voimakasasteinen takaisinvirtaus liittyy osana munuaisen pienikokoisuuteen johtavaan kehityshäiriöön eikä siten ole munuaisvaurion aiheuttaja. Sen sijaan leikki- ja kouluikäisillä tytöillä virtsan takaisinvirtaus yhdessä toistuvien pyelonefriittien kanssa voi johtaa munuaisen kuorikerroksen vaurioitumiseen (6).
Vaikeisiin synnynnäisiin virtsaelinten kehityshäiriöihin liittyy pienentynyt lapsiveden määrä ja sen seurauksena sikiön keuhkojen normaali kehitys häiriintyy. Nämä lapset ovat syntyessään vaikeasti sairaita ja tarvitsevat tehohoitoa. Imeväisillä ja leikki-ikäisillä virtsaelinten kehityshäiriöihin liittyvä oireisto voi olla varsin vaihteleva tai oireita ei ole lainkaan ja poikkeavuus löytyy sattumalta. Lapsella voi olla toistuvia virtsatietulehduksia, runsasvirtsaisuutta ja jatkuvaa janon tunnetta, kasvu voi olla hidastunut tai verenpaine koholla.
Virtsaelinten rakennepoikkeavuuksien selvittely kannattaa aloittaa kaikututkimuksella, koska sillä voidaan varsin luotettavasti sulkea pois merkittävät kehityshäiriöt. Jatkokuvantamisten tarpeesta on hyvä neuvotella lastennefrologin tai -urologin kanssa.
Munuaisten ja virtsaelinten synnynnäisten kehityshäiriöiden pitkäaikaisennuste vaihtelee lähes oireettomasta vaikeaan munuaisten vajaatoimintaan. Suomessa lasten munuaisensiirtoihin johtavista sairauksista synnynnäiset virtsaelinten kehityshäiriöt ovat toiseksi suurin diagnoosiryhmä (noin 25 %). Pohjoismaisen lasten munuaisensiirtorekisterin mukaan osuus on pysynyt samana vuodesta 1986 lähtien (7).
Lapsia, joilla on todettu virtsaelinten kehityshäiriö, suositellaan seurattavaksi harvakseltaan, ainakin kolmen vuoden välein. Tällä varmistetaan normaali kasvu sekä munuaisten vajaatoiminnan tai verenpainetaudin mahdollisimman varhainen toteaminen.
Munuaisten rakkulasairaudet
Munuaisrakkulat ovat munuaisten yleisin rakenteellinen poikkeavuus. Ne voidaan havaita ensimmäisen kerran jo raskaudenaikaisessa sikiön rakenneultraäänitutkimuksessa tai sattumalöydöksenä vatsan kaikututkimuksen yhteydessä.
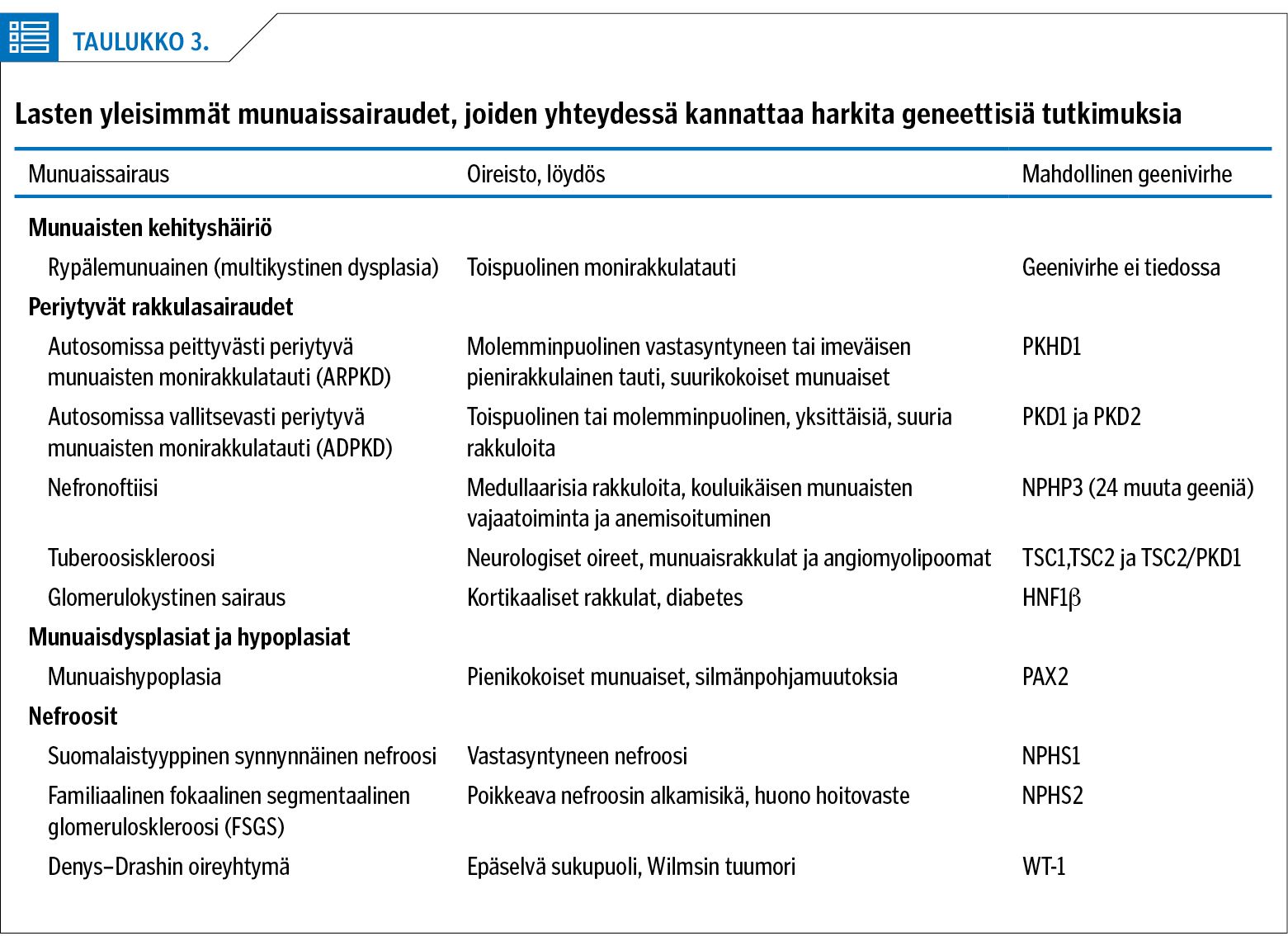
Munuaisrakkuloita voi liittyä useisiin tautitiloihin (taulukko 3). Rypälemunuainen eli multikystinen dysplasia luokitellaan munuaisten kehityshäiriöksi. Tunnettuja geenivirheiden aiheuttamia munuaisten rakkulasairauksia ovat autosomissa peittyvästi (ARPKD) ja vallitsevasti (ADPKD) periytyvä munuaisten monirakkulatauti sekä nefronoftiisit. Harvinaisempia ovat glomerulokystiset sairaudet, jotka voivat liittyä tuberoosiskleroosiin (TSC1-2/PKD1 -mutaatiot) (8).
Ymmärrys munuaisten rakkulasairauksien syntymekanismeista ja sairauksien diagnostiikka ovat tarkentuneet uusien genetiikan tutkimusmenetelmien avulla. Rakkulasairaudet liittyvät värekarvojen toimintahäiriöihin. Niihin voi liittyä munuaisten vajaatoiminnan lisäksi maksan ja haiman toimintahäiriöitä sekä kuulonalenemaa ja neurologisia oireita.
Rypälemunuaisen ilmaantuvuus on noin yksi tapaus 4 000–5 000 elävänä syntynyttä kohden (8). Tila on yleensä toispuolinen, mutta 20–30 %:lla potilaista toisessa munuaisessa on jokin rakennepoikkeavuus, kuten virtsan takaisinvirtaus. Rypälemunuaisen ei tiedetä liittyvän yhden geenin mutaatioon.
Autosomissa peittyvästi periytyvä monirakkulatauti ARPKD aiheutuu mutaatioista PKHD1-geenissä. Sen yleisyys eri väestöissä on 1/10 000–40 000 elävänä syntynyttä. Taudin vaikeassa muodossa vastasyntyneen munuaiset ovat hyvin suuret ja lapsella on keuhkohypoplasia. Koska parantavaa hoitoa ei ole, lievemmissäkin muodoissa tauti etenee munuaisten loppuvaiheen vajaatoimintaan tyypillisesti alle 20 vuoden iässä ja noin puolet potilaista joutuu munuaisensiirtoon alle 10-vuotiaina (8). Maksafibroosin kehittyminen on hyvin vaihtelevaa ja osa potilaista tarvitsee myös maksansiirron. Suomessa on kahdeksalle lapselle tehty yhdistetty munuaisen- ja maksansiirto ARPKD-taudin vuoksi.
Vallitsevasti periytyvän monirakkulataudin ADPKD:n aiheuttavat mutaatiot PKD1- ja PKD2-geeneissä. Se on peittyvästi periytyvää tautia huomattavasti yleisempi (1/400–1 000) (8). ADPKD-potilailla munuaisten vajaatoiminta kehittyy selvästi myöhemmin: potilaista, joilla on PKD1-mutaatio, puolet on dialyysihoidossa noin 50-vuotiaina, ja PKD2-mutaatio johtaa dialyysihoitoon keskimäärin 70 vuoden iässä (8). Taudin hoitoon on aikuisilla käytetty rakkuloiden kasvua hidastavaa antidiureettisen hormonin estäjää, tolvaptaania. Lapsilla satunnaistettuja tutkimuksia ei ole tehty. Nykyhoito perustuu munuaisten vajaatoiminnan lääkkeelliseen tukihoitoon sekä tyypillisen liitännäissairauden, korkean verenpaineen hoitoon.
Medullaariset rakkulasairaudet, nefronoftiisit, on yhdistetty NPHN-geenin mutaatioihin, mutta myöhemmin on todettu, että saman ilmiasun aiheuttavia mutaatioita ilmenee yli 25 geenissä (9). Merkittävä osa nefronoftiisipotilaista tarvitsee munuaisensiirron. Sairauden etenemisnopeuteen vaikuttaa geenivirheen laatu, joskin kaikkien rakkulasairauksien genotyypin ja ilmiasun välinen korrelaatio on melko huono.
Munuaiskerässairaudet
Nefroottinen oireyhtymä tarkoittaa runsasta valkuaisaineiden erittymistä virtsaan (U-Prot/krea-suhde yli 200 mg/mmol) ja sitä seuraavia veren valkuaisainepitoisuuden pienenemistä ja turvotuksia (10). Erillisen nefroottistasoisen proteinurian taustalla on yleisimmin idiopaattinen nefroottinen oireyhtymä. Tämän tyypillisimmin leikki- ja kouluikäisillä esiintyvän sairauden syntymekanismi on epäselvä, mutta sen on arveltu olevan munuaiskerästen mikrokapillaarien epiteelisolujen, podosyyttien, toimintahäiriö. Hoitovaste prednisoloniin sekä kalsineuriinin estäjiin (siklosporiini A ja takrolimuusi) on usein hyvä, mikä viittaa immunologiseen patomekanismiiin. Nefroottisen oireyhtymän uusiutumisriski vähenee merkittävästi murrosiän jälkeen ja nefroosin pitkäaikaisennuste on hyvä (11).
Nefroottinen oireyhtymä ja proteinuria voivat aiheutua munuaiskerästen kapillaarien rakennemolekyylien geneettisistä sairauksista, joista tunnetuin on suomalaistyyppinen synnynnäinen nefroosi (CNF). Sen aiheuttaa mutaatio nefriiniä koodaavassa geenissä (NPHS1) (12). CNF-lapset syntyvät yleensä hieman ennenaikaisina, istukan koko on suurentunut ja lapsella todetaan turvotuksia sekä massiivinen proteinuria (12). Taudin yleisyys on suomalaisessa kantaväestössä noin 1/8 000 synnytystä ja uusia tautitapauksia todetaan 2–4 vuodessa. Muita synnynnäistä nefroosia aiheuttavia geenivirheitä on kuvattu muun muassa NPHS2-, LAMB2-, PLCE1- ja WT-1-geeneissä (10).
Toisin kuin nefrooseissa yleensä, imeväisten tautimuodoissa ennuste on huono, ja merkittävä osa näistä potilaista päätyy munuaisensiirtoon. Geenivirheen aiheuttamaan tautiin viittaavat proteinurian ilmaantuminen vastasyntyneelle tai murrosikäisille nuorille, huono vaste immunosuppressiiviseen hoitoon ja suvussa esiintyvä proteinuria. Näissä tapauksissa onkin syytä harkita geneettisiä tutkimuksia.
Munuaiskerästen tulehdukset, glomerulonefriitit, ovat lapsilla varsin harvinaisia. Niille tyypillistä on veri- ja valkuaisvirtsaisuus sekä hankalammissa tapauksissa nefriittiseen oireyhtymään liittyvät muut löydökset, kuten munuaisten vajaatoiminta (suurentunut plasman kreatiniini- ja kystatiini C -pitoisuus), vähentynyt virtsaneritys, turvotukset ja poikkeavan korkeat verenpaineet. Munuaiskerästulehdus voi hoitamattomana edetä nopeasti merkittävään munuaisten vajaatoimintaan, ja tämän vuoksi nämä potilaat on syytä lähettää nopeasti lastennefrologin arvioon.
Yleisimmät lasten munuaiskerästulehdukset ovat Henoch–Schönleinin purppuraan liittyvä nefriitti (HSN), IgA-nefropatia ja useimmiten A-streptokokkitulehduksiin liittyvä infektionjälkeinen proliferatiivinen munuaiskerästulehdus. Sekä Henoch–Schönleinin purppura että infektionjälkeinen glomerulonefriitti paranevat useimmiten ilman hoitoa. Vaikeita Henoch–Schönlein-tapauksia hoidetaan Suomessa yleisimmin siklosporiinilla (13). Harvinaisempia munuaiskerästulehdukseen johtavia sairauksia ovat ANCA-vaskuliitit (mikroskooppinen polyangiitti ja granulomatoottinen polyangiitti) sekä systeeminen lupus erythematosus (SLE) (2,13.14).
Munuaiskerästulehdusten ennuste on parantunut merkittävästi muun muassa biologisten lääkkeiden tultua markkinoille. Pohjoismaisen lasten munuaisensiirtorekisterin mukaan munuaiskerästulehdusten vuoksi tehtävien munuaisensiirtojen osuus on pienentynyt noin 10 % verrattaessa lapsille vuosina 1986–1996 ja 1997–2012 tehtyjen munuaissiirtojen syitä (7,15).
Hemolyyttis-ureemisessa oireyhtymässä (HUS) munuaiskerästen kapillaareihin muodostuu mikrotrombeja bakteeritoksiinien (enterohemorraginen E. coli, EHEC) tai komplementin säätelyyn osallistuvien tekijöiden häiriintyneen toiminnan vuoksi. Tämä aiheuttaa taudin tyypilliset oireet: hemolyyttisen anemian, trombosytopenian ja munuaisten vajaatoiminnan. Osalla potilaista esiintyy myös keskushermosto-oireita (16). Komplementin säätelyhäiriöön voi johtaa komplementin tekijöiden geneettinen poikkeavuus tai niitä vastaan muodostuneet vasta-aineet (17). Bakteeri-infektioon liittyvä HUS paranee yleensä itsestään. Sen sijaan epätyypillinen HUS hoidetaan tarvittaessa komplementin tekijä C5:n vasta-aineella, ekulitsumabilla (17).
Alportin oireyhtymä on perinnöllinen munuaiskerässairaus. Sen aiheuttaa joko X-kromosomissa tai autosomissa peittyvästi periytyvä kollageenin rakennepoikkeavuus, joka johtaa munuaiskerästen mikrokapillaarien tyvikalvon poikkeavuuteen ja veri- ja valkuaisvirtsaisuuteen sekä munuaisten vajaatoimintaan (18). Alportin oireyhtymään liittyy tyypillisesti kuulonalenema. Sairauden vaikeusaste vaihtelee sukupuolen ja perityn mutaation perusteella. Oireisto ilmenee yleensä kouluikäisillä lapsilla (19).
Munuaistiehyiden ja välikudoksen sairaudet
Munuaistiehyiden eli tubulusten sairaudet ovat useimmiten geneettisiä elektrolyyttitasapainon säätelyhäiriöitä tai esimerkiksi syövän hoitojen aiheuttamia toksisia munuaistiehyiden toimintahäiriöitä. Perinnöllisistä sairauksista yleisimmin tavattavia ovat Bartterin oireyhtymä (hypokaleeminen alkaloosi), Gitelmanin oireyhtymä (hypokalemia ja hypomagnesemia), renaalinen tubulaarinen asidoosi sekä nefrogeeninen diabetes insipidus (20). Tubulussairaudet on syytä pitää mielessä mietittäessä syitä elektrolyyttitasapainon häiriöihin, poikkeavaan asidoosiin tai alkaloosiin, imeväisen huonoon menestymiseen tai lapsen kasvun hidastumiseen.
Munuaisvälikudoksen sairauksista yleisin on pyelonefriitti. Muita harvinaisempi sairauksia ovat Puumala-viruksen aiheuttama myyräkuume sekä idiopaattinen tubulointerstitiaalinefriitti (19,21). Munuaisvälikudoksen tulehdussairauksiin liittyvät tyypillisesti nefriitin kuva, suurentuneet tulehdusarvot, anemisoituminen sekä pienimolekyylipainoisten valkuaisaineiden ja glukoosin poikkeava erittyminen virtsaan. Interstitiaaliseen nefriittiin sairastuneilla nuorisoikäisillä potilailla todetaan usein uveiitti (22).
Diagnostiset mahdollisuudet ja hoitokäytännöt
Lasten munuaissairauksien diagnostiikka perustuu pitkälti kuvantamistutkimuksiin, munuaistoimintaa mittaavien merkkiaineiden, kuten plasman kreatiniinin ja kystatiini C:n, pitoisuuksien mittaamiseen, serologisiin tutkimuksiin, virtsa-analyyseihin, histologisiin tutkimuksiin sekä geneettisiin tutkimuksiin.
Merkittävin muutos diagnostiikassa on uuden sukupolven geenisekvensointimenetelmien tulo kaupalliseen käyttöön. Nämä suorasekvensointimenetelmät ovat nopeuttaneet diagnostiikkaa ja mahdollistaneet usean geenin samanaikaisen sekvensoinnin. Geeniteknologian kehittymisen ansiosta munuaissairauksien diagnostiikka on tarkentunut, ja mutaatioanalyysit ovat vähentäneet usein anestesiaa vaativaa munuaiskoepalojen ottamista. Esimerkiksi Alportin tautia tai nefronoftiisia epäiltäessä diagnoosi voidaan tehdä pelkän geenitutkimuksen avulla.. Lisäksi lähiomaisten tutkimus- ja seurantatarvetta voidaan kohdentaa paremmin selvittämällä aiheuttajamutaation esiintyvyys perheessä.
Uudet biologiset lääkkeet (rituksimabi, ekulitsumabi) ovat parantaneet erityisesti vasta-ainevälitteisten ja komplementin toimintahäiriöihin liittyvien munuaissairauksien ennustetta. Kalsineuriinin estäjien ja puriininukleotidien estäjän mykofenolihapon lisääntynyt käyttö munuaissairauksien hoidossa on myös osaltaan vaikuttanut pitkäaikaisennusteeseen suotuisasti. Toisaalta tiedetään, että nämä lääkkeet vaikuttavat elimistön puolustusmekanismeihin ja altistavat erityisesti herpesryhmän virusinfektioille. Lisäksi ne voivat pitkäaikaisessa käytössä aiheuttaa munuaisten vajaatoimintaa ja lisätä syöpäriskiä. Lääkityksen aloittaminen, lääkehoidon vasteen seuranta ja hoidon keston suunnittelu kuuluvat hoitoon erikoistuneille asiantuntijoille.
Munuaisensiirto on vakiintunut parantumattoman munuaisten loppuvaiheen vajaatoiminnan hoitomuoto lapsilla. Lasten munuaisensiirrot on keskitetty HUS:n Uuteen lastensairaalaan. Munuaisensiirto voidaan tehdä noin kymmenkiloiselle lapselle. Noin puolet lasten munuaissiirrännäisistä saadaan eläviltä luovuttajilta, yleisimmin jommaltakummalta vanhemmalta. Siirroista 20–30 % tehdään ennakoivasti, ilman edeltävää dialyysihoitoa. Suomessa munuaissiirteen saaneiden lasten eloonjäämisennuste on noin 95 % ja siirteen toimintaennuste kymmenen vuoden kuluttua siirrosta on noin 90 %. Potilaat tarvitsevat elinikäistä hyljinnänestolääkitystä ja pitkäaikaisseurantaa.
Osa lapsena munuaissairauteen sairastuneista aikuistuvista potilaista tarvitsee nefrologin seurantaa, osalle potilaista opiskelijaterveydenhuollon, työterveyshuollon tai terveyskeskuksen seuranta on riittävä. Lapsuudessa munuaissairautta sairastaneiden naisten raskaudet edellyttävät tarkkaa seurantaa lisääntyneen pre-eklampsiariskin vuoksi (19). Aikuistuville munuaissairautta sairastaville nuorille on tärkeää laatia yksilöllinen suunnitelma seurannan tarpeesta ja järjestää seuranta yhdessä jatkohoitopaikan kanssa.
Lopuksi
Suurin osa lasten munuaissairauksista on ennusteeltaan hyvänlaatuisia. Myös lasten vaikeiden munuaissairauksien ennuste on parantunut diagnostisten menetelmien ja lääkehoidon kehittymisen ansiosta. Vaikean munuaisten vajaatoiminnan vuoksi munuaiskorvaushoitoon päätyneiden lasten ja nuorten eloonjäämisennuste on hyvä. Hyvien hoitotulosten jatkuminen edellyttää korkealaatuisen tutkimustoiminnan ja ammatillisen osaamisen ylläpitämistä.
Timo Jahnukainen: Ei sidonnaisuuksia.
- 1
- Suomen munuaistautirekisteri. Vuosiraportit. https://www.muma.fi/liitto/suomen_munuaistautirekisteri
- 2
- Jalanko H, Rönnholm K, Holmberg C. Lasten vaikeat munuaissairaudet. Duodecim 1997;113:645–53.
- 3
- Jahnukainen T, Rönnholm K. Veri- ja valkuaisvirtsaisuus lapsella. Suom Lääril 2015;70:1149–55.
- 4
- Grandjean H1, Larroque D, Levi S. The performance of routine ultrasonographic screening of pregnancies in the Eurofetus Study. Am J Obstet Gynecol 1999;181:446–54.
- 5
- Uy N, Reidly G. Developmental genetics and congenital anomalies of the kidney and urinary tract. J Pediatr Genet 2016;5:51–60.
- 6
- Mattoo TK, Chesney RW, Greenfield SP ym. Renal scarring in the Randomized Intervention for Children with Vesicoureteral Reflux (RIVUR) Trial. Clin J Am Soc Nephrol 2016;11:54–61.
- 7
- Jahnukainen T, Bjerre A, Larsson M ym. The second report of the Nordis Pediatric Renal Transplantation Registry 1997-2012: More infant recipients and improved graft survival. Pediatr Transplant 2016;20:364–71.
- 8
- Kwatra S, Krishnappa V, Mhanna C ym. Cystic diseases of childhood: A review. Urology 2017;110:184–91.
- 9
- Luo F, Tao Y-H. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology 2018;23:904–11.
- 10
- Wang C-S, Greenbaum LA. Nephrotic syndrome. Pediatr Clin N Am 2019;66:73–85.
- 11
- Lahdenkari AT, Suvanto M, Kajantie E, Koskimies O, Kestilä M, Jalanko H. Clinical features and outcome of childhood minimal change nephrotic syndrome: is genetics involved? Pediatr Nephrol 2005;20:1073–80.
- 12
- Holmberg C, Jalanko H. Suomalaistyyppinen synnynnäinen nefroosi – avain proteinurian mekanismeihin. Duodecim 2011;127:1017–25.
- 13
- Ronkainen J, Koskimies O, Ala-Houhala M ym. Henoch-Schönleinin purppura lapsilla. Duodecim 2017;123:1329–37.
- 14
- Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front Pediatr 2018;6:226. doi: 10.3389/fped.2018.00226.
- 15
- Tydén G, Berg U. Pediatric renal transplantation in the Nordic countries: a report of the Nordic Renal Transplant Study Group. Pediatr Transplant 1998;2:240–3.
- 16
- Korhonen L, Nuutinen M. EHEC-infektiot ja lasten hemolyyttis-ureeminen oireyhtymä OYS:ssa 1995–2016. Suom Lääkäril 2019;74:1427–31.
- 17
- Kaartinen K, Martola L, Meri S. Epätyypillinen hemolyyttis-ureeminen oireyhtymä. Duodecim 2017;133:539–47.
- 18
- Savige J, Gregory M, Gross O ym. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 2013;24:364–75.
- 19
- Syrjänen T, Mustonen J, Vapalahti O, Henttonen H, Vaheri A. Jyrsijöiden levittämät taudit Suomessa. Duodecim 2005;121:295–302.
- 20
- Seyberth HW. Pathophysiology and clinical presentation of salt-losing tubulopathies. Pediatr Nephrol 2016;31:407–18.
- 21
- Jahnukainen T, Ala-Houhala M, Saarela V, Nuutinen M. Lasten akuutti tubulointerstitaalinefriitti. Duodecim 2005;121:998–1000.
- 22
- Ronkainen J, Ala-Houhala M, Huttunen NP ym. Long-term outcome 19 years after childhood IgA nephritis: a retrospective cohort study. Pediatr Nephrol 2006;21:1266–73.
Kidney diseases in children
Kidney diseases are relatively common among children. They may be caused by congenital abnormalities affecting the kidneys or urinary tract, glomerular or tubular disorders. The symptoms of kidneys diseases are variable. Renal insufficiency is an important differential diagnostic option when examining a child with growth failure, failure to thrive, or hypertension.
The incidence of children with end stage kidney disease (ESRD) has remained stable, however, the causes of ESRD have changed during recent years. The survival of children with ESRD has improved and the number of patients with a history of severe kidney disease during childhood who will be transferred to adult care is increasing.






{kind=link}
{kind=link}
{kind=link}