Missä viipyvät Alzheimerin taudin uudet lääkkeet?
Alzheimerin tautiin on toistaiseksi onnistuttu kehittämään ainoastaan oireita lieventäviä hoitoja. Niistä uusin on otettu käyttöön 2003.
Uusien lääkkeiden kehitys on viime vuosikymmenen aikana keskittynyt amyloidiproteiineista muodostuneiden peptidiplakkien muokkaamiseen, mutta lukuisat faasin III tutkimukset ovat epäonnistuneet.
Tutkimusten perusteella hoito tulisi aloittaa jo ennen kognitiivisten muutosten alkua.
Tulevaisuudessa uudet vaikutusmekanismit ja diagnostiset merkkiaineet auttavat potilaiden varhaista tunnistamista ja ehkäisevää hoitoa.

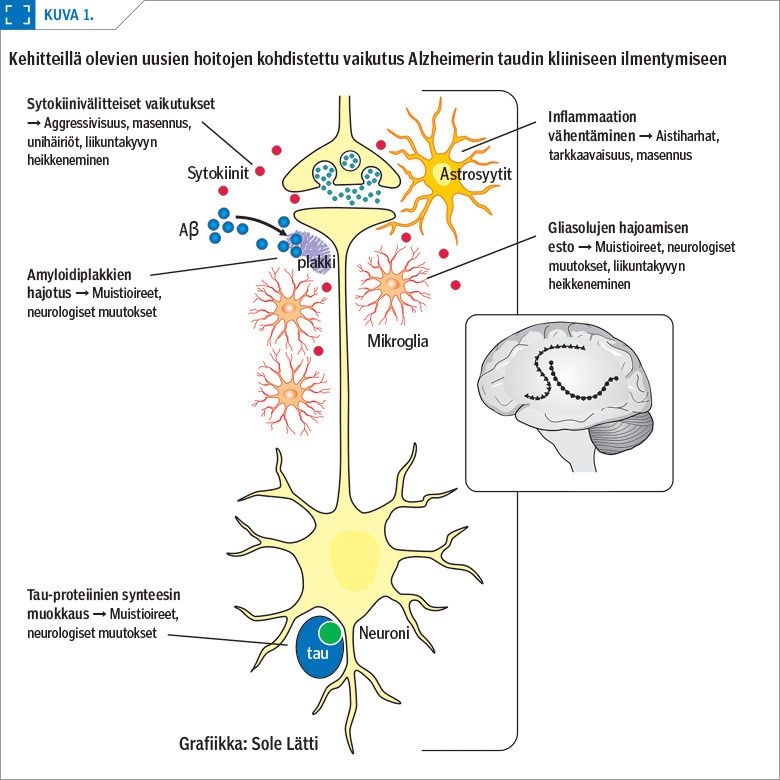

Alzheimerin tauti luokitellaan nykyisellään parantumattomaksi. Sen merkittävimpiin neuropatologisiin muutoksiin kuuluvat amyloidiproteiineista koostuvien peptidiplakkien muodostus, neurofibrillaariset tau-proteeineista koostuvat kimput (neurofibrillary tangles), hermosolujen välisten yhteyksien katkeaminen ja lopulta yleistyvä aivojen atrofia (1).
Nykytietämyksen mukaan amyloidiplakit ja tau-proteiinikimput alkavat muodostua jopa yli kymmenen vuotta aikaisemmin kuin havaittavissa ja mitattavissa olevat kognitiiviset muutokset (2). Alzheimerin taudin lääkekehitystä onkin viime vuosikymmenen ajan hallinnut vahva keskittyminen nimenomaan amyloidiplakkien muodostumisen ehkäisyyn tai tuhoamiseen.
Toistaiseksi hyväksyttyjä lääkkeitä on saatavilla ainoastaan oireiden lievitykseen, eikä yhtään niistä ole luokiteltu sairautta parantavaksi lääkkeeksi. Lääkkeiden hyväksynnästä on kulunut jo tovi: donepetsiili 1997, rivastigmiini 2000, galantamiini 2001 ja memantiini 2003 (3). Lääketeollisuutta ei voida kuitenkaan syyttää yrittämisen puutteesta, sillä viime vuosikymmenten aikana useita uusia lääkemolekyylejä on tutkittu kliinisesti varsin pitkälle, jopa faasin III tutkimuksiin saakka.
Eri lähteiden perusteella kliinisesti tutkittuja molekyylejä on ollut 100–150 (3,4), sen mukaan, lasketaanko erilaiset taudin esiasteet ja niissä tutkitut molekyylit mukaan. Alzheimerin taudin lääkehoitoon tähtäävät tutkimukset ovat kuitenkin kerta toisensa jälkeen epäonnistuneet. Mistä tämä voi johtua?
Haasteelliset amyloidipeptidit
Alzheimerin taudin syytä ei edelleenkään tunneta. Viime vuosikymmenten epäonnistuneita lääketutkimuksia on yhdistänyt vahva keskittyminen ns. amyloidihypoteesiin. Vaikutusmekanismit ovat olleet suunnattuja amyloidipeptideistä koostuvien plakkien muodostumisen estämiseen tai jo muodostuneiden plakkien hajottamiseen. Vielä ennen vuotta 2019 käynnistyneistä lääketutkimuksista reilusti yli puolet piti amyloidipeptidejä kohdemolekyyleinään (3,4).
Amyloidihypoteesi juontaa juurensa geenitutkimuksissa tehtyihin löydöksiin. Alzheimerin taudissa tiedetään olevan geenivirheitä, jotka ilmetessään aina aiheuttavat sairauden. Tällaisia mutaatioita tunnetaan kolmesta geenistä: kromosomissa 21 olevassa amyloidin prekursoriproteiinia (APP) koodaavassa geenissä, kromosomin 14 preseniliini 1 (PSEN1) -geenissä ja kromosomin 1 preseniliini 2 (PSEN2) -geenissä (5,6,7,8). Kustakin geenistä tunnetaan useita mutaatioita, ja ne kaikki edistävät amyloidiplakkien kertymistä aivoihin.
Mutaatiot aiheuttavat Alzheimerin taudin ehkäisemällä amyloidipeptidien normaalia pilkkoutumista kahdella mahdollisella tavalla: joko itse proteiini on viallista (APP-mutaatiot) tai sitä pilkkovat entsyymit ovat viallisia (PSEN 1- ja 2-mutaatiot). Amyloidiplakkien merkkiaineiden tai niiden prekursorien määrittäminen selkäydinnesteestä onkin toiminut erilaisten kuvantamismenetelmien lisäksi yhtenä keskeisenä diagnostisena työkaluna varsinkin varhaisen vaiheen sairauden toteamisessa.
Amyloidiproteiineilla on nykytietämyksen mukaan useita fysiologisia funktioita. Niitä voidaan osoittaa laajalti aivoista ja muualta kehosta niin terveiltä kuin muistisairailtakin henkilöiltä (9). Liukoiset amyloidiproteiinit ovat osallisina monissa fysiologisissa toiminnoissa, esimerkiksi synapsien toiminnan modulaatiossa ja neuronien kasvun stimulaatiossa (10).
APP:n pilkkoutuminen viallisiksi beeta-amyloidipeptideiksi (Aβ) ja peptidien kertyminen plakkimaisiksi rakenteiksi edistävät inflammaatiotilan muodostumista. Monet amyloidiproteiinit toimivat inflammaatiota edistävinä yhdisteinä, jotka aktivoivat monia kehon inflammatorisia järjestelmiä (11). Alzheimerin taudin varhaisissa vaiheissa gliasolujen aktivaatio voi olla merkki suojamekanismista, jossa solut yrittävät hajottaa amyloidiplakit ja vahvistaa hermosolujen kasvutekijöitä.
Kuvantamismenetelmillä on pystytty havaitsemaan, että amyloidiplakkien muodostuminen on todennäköisesti alkanut paljon aiemmin kuin taudin dementiavaiheeseen liittyvää aivojen atrofiaa on havaittavissa. Kun amyloidiplakkia on muodostunut runsaasti, on jo usein määritettävissä varhaisia kognitiivisia oireita. Kun plakkien määrä kasvaa haitallisen suureksi, alkavat useat inflammaatiota edistävät järjestelmät käynnistyä, jolloin myös neuronit vahingoittuvat (10,11). Havaintoja tällaisista mekanismeista on myös todettu Alzheimerin tautiin kuolleiden potilaiden näytteissä (12,13).
Viime vuosikymmenen aikana myös perifeeristen inflammatoristen prosessien on osoitettu olevan keskeisessä roolissa taudin patologiassa, ja tämä on heijastunut myös inflammatorisia prosesseja kohteena hyödyntävien lääkkeiden kehityksessä. Teorian mukaan perifeerinen inflammaatio indusoi sentraalisia inflammatorisia mekanismeja ja edelleen Aβ:n kasautumista, mikä puolestaan aloittaa monia uusia inflammatorisia prosesseja. Vielä ei kuitenkaan ole pystytty osoittamaan, onko primaarinen inflammatorinen prosessi lähtöisin amyloidiproteiineista vai ovatko amyloidit seurausta perifeerisestä inflammaatiosta (11).
Tulevaisuuden tau
Toisena keskeisenä lääketutkimusten kohteena ovat viime vuosina olleet tau-proteenien aiheuttamat ns. proteiinivyyhdet tai neurofibrillikimput. Tau-proteiinien kertyminen voi olla seurausta neuronien kokemista vahingollisista muutoksista (14).
Tutkimuksissa on saatu näyttöä, että Alzheimerin taudin kehitys voisi alkaa amyloidiplakkien kertymisestä, mutta jossain vaiheessa taudin kulkua myös tau-proteiinit alkavat muodostaa kimppuja (15). Tau-proteiinista muodostuneilla kimpuilla onkin osoitettu paremmin korreloiva yhteys taudin vakavuuteen ja dementiavaiheen kestoon, ja niitä tai tau-proteiinin fosforyloitunutta muotoa on käytetty viime vuodet yhtenä tärkeimmästä biomarkkereista taudin diagnostiikassa (16). Tau-proteiinikimppujen kehittyminen ei sinällään edellytä amyloidiplakkien läsnäoloa, koska esimerkiksi monissa iskeemisissä tai traumaattisissa sairauksissa kimppuja voidaan tavata ilman merkkejä edeltävästä amyloidikertymästä (17).
Tau-proteiinit ovat kiistatta yhteydessä hermosolutuhon etenemiseen ja toimivat epäspesifisinä tuhoutumista ennakoivina ja sen jälkeen ilmenevinä aksonivaurion merkkeinä, kun taas fosforyloitunutta tau-proteiinia pidetään spesifisempänä diagnostisena markkerina nimenomaan Alzheimerin taudille (17). Lisäksi PET-tutkimuksilla on osoitettu tau-proteiinimuutosten korreloivan Alzheimerin taudin kliinisiin oireisiin merkittävästi paremmin kuin amyloidiplakkien muutokset (14,15,16,17). Nykyään vahvimman diagnostisen kokonaisuuden muodostaa erilaisten neurodegeneraatiota määrittävien kuvantamismenetelmien yhdistäminen amyloidipeptidien ja tau-proteiinien määriin (18).
Kliiniset lääketutkimukset 2020
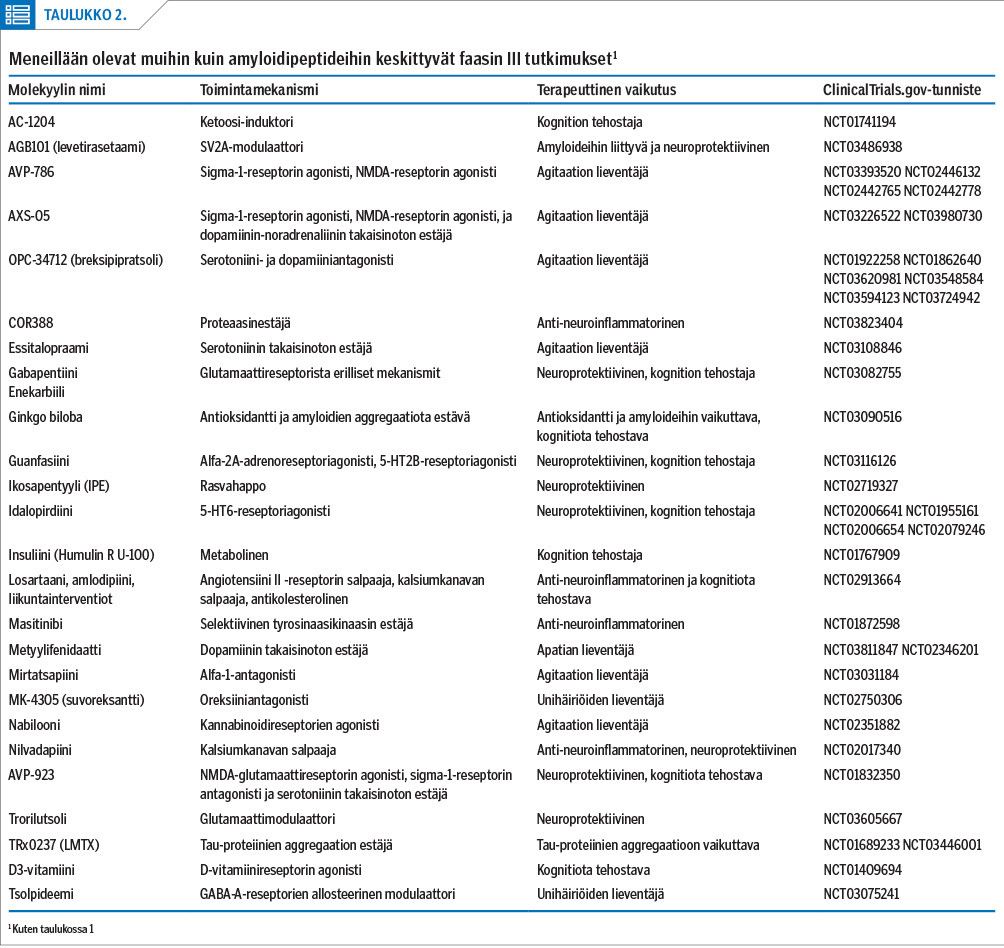
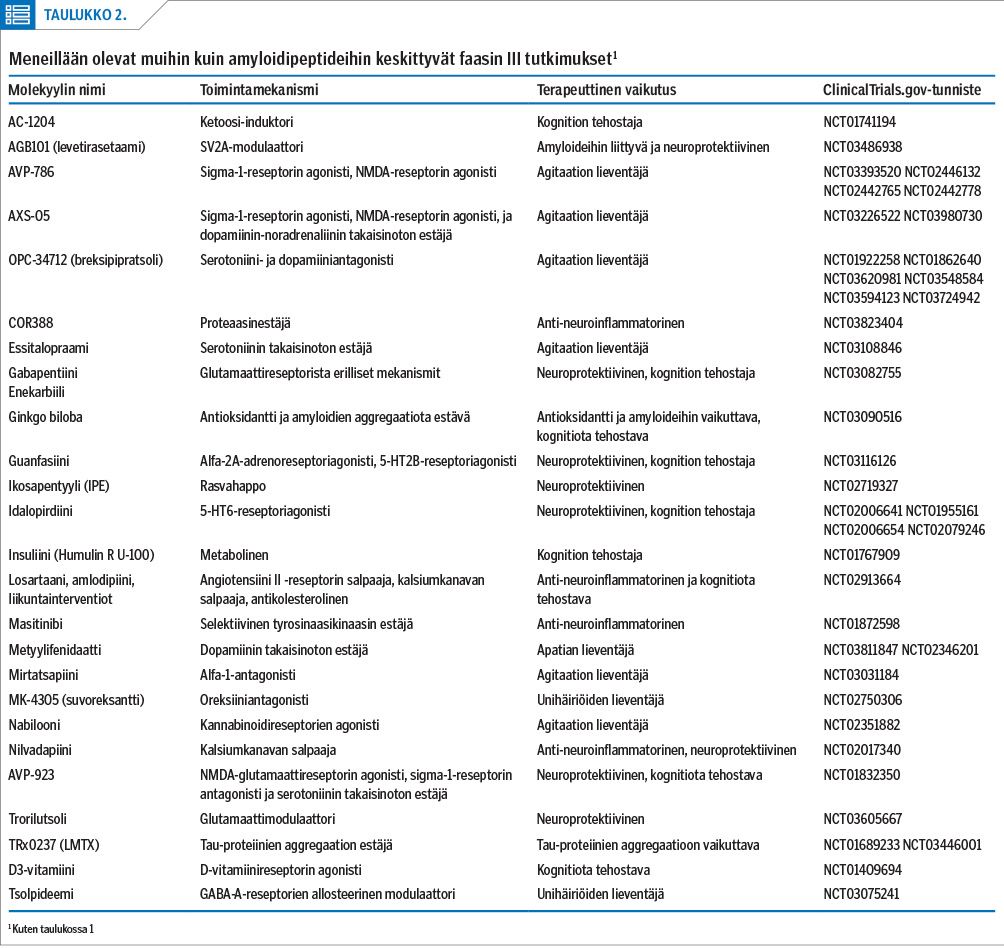
Tätä kirjoittaessa vielä avoimina olevia kliinisiä Alzheimerin tautiin liittyviä faasin III lääketutkimuksia on Clinicaltrials.gov-sivuston (haku suoritettu 4.11.2020) perusteella noin viisikymmentä. Näistä amyloidiproteiineihin kohdennettuja tutkimuksia on enää vain 12, ja hieman alle neljässäkymmenessä tutkimuksessa kohteena on jokin muu lääkemekanismi (kuva 1). Samasta tietokannasta löytyy kahdeksan eri amyloidiproteiineja kohdentavaa molekyyliä (taulukko 1).
Kahdessa tutkimuksessa tutkitaan prekliinisen tautivaiheen potilaita. Toisessa edellytettiin positiivinen PET-kuvantamistulos amyloidien suhteen, toisessa geneettisiä riskitekijöitä tai todennettuja geenimuunnoksia. Neljässä tutkimuksessa rekrytoitiin prodromaali- eli varhaisen vaiheen tai prekliinisen eli oireettoman vaiheen Alzheimer-potilaita. Kaikissa edellytettiin positiivinen biomarkkerilöydös ja myös positiivinen PET-kuvantamisen tulos.
Biomarkkereina näissä tutkimuksissa käytettiin sekä selkäydinnesteen tau-proteiinia että amyloidipeptidejä. Lisäksi joskus mainitaan ehdollisena löydöksenä suureen tautiriskiin yhdistetty mutaatiolöydös APOE-geenin E4-alleelissa. Tautivaiheen määrityksessä käytettiin uudempia NIA-AA (National Institute of Aging Alzheimer’s Association), MCI (Mild Cognitive Impairment) tai MCI-AD (Mild Cognitive Impairment in Alzheimer’s Diseases) -kriteerejä.
Edenneen Alzheimerin taudin tutkimuksia ei enää tietokannasta löytynyt. Näin ei ollut vielä kolme vuotta sitten (3).
Muita hoidon kohteita on kuitenkin tullut amyloidiproteiinien tilalle (taulukko 2). Tietokannan mukaan kolme molekyyliä on faasin III tutkimuksissa kohdennettu tau-proteiineihin. Neuroinflammaation lieventäjiä löytyy samassa vaiheessa kaksi, neuroprotektiivisia molekyylejä kolme, kognition tehostajia kaksi ja potilaiden käytösoireisiin vaikuttavia lääkkeitä ainakin neljä. Lisäksi osalla molekyyleistä uskotaan olevan useita yhtäaikaisia vaikutusmekanismeja. Uusia tutkimuksia syntyy siten edelleen.
Loistava tulevaisuus
Tiedeyhteisön käsityksen perusteella amyloidiplakkien voidaan lähtökohtaisesti ajatella olevan hyvä kohde uusille lääkemolekyyleille. Faasin III tutkimusten epäonnistumiselle on osoitettu useita syitä. Tärkeimpänä pidetään liian myöhäistä hoidon aloittamista (3,4,13,18).
Tutkimukset ovatkin viime aikoina perustellusti keskittyneet prodromaalisen tai prekliinisen Alzheimerin taudin hoitoon. Näissä haasteelliseksi on muodostunut se, että hyväksyttyjen hoitojen puuttuessa verrattain kalliille diagnostiikalle ei ole ollut kliinisiä perusteita. Tämä on vaikeuttanut myös tutkimusten rekrytointiprosessia ja sopivien potilaiden tunnistamista (4).
Määritelmän mukaan prodromaalisessa Alzheimerin taudissa biomarkkerit viittaavat alkavaan sairauteen, mutta kliinisesti potilaalla on todettu vain lieviä kognition muutoksia, eikä tilanne siten täytä Alzheimerin taudin dementiavaiheen diagnostisia kriteereitä (18). Prekliinisen eli kliinisesti oireettoman tautivaiheen määrittämiseksi puolestaan käytetään useita erilaisia diagnostisia biomarkkereita (4,18), eikä sen toteamiseen vaadita muita kriteerejä. Dubois ym. ovat koonneet varhaisen Alzheimerin taudin kriteerit kattavassa katsausartikkelissaan (19), mutta näitä kriteerejä on toistaiseksi hyödynnetty kliinisissä lääketutkimuksissa verrattain vähän.
Aiemmista tutkimuksista on myös opittu, että tilanteessa, jossa potilaalla on huomattu lieviä muutoksia kognitiossa, lääkehoidon aloittaminen voi olla jo liian myöhäistä. Usein tässä vaiheessa amyloidiplakit ovat jo vahingoittaneet aivoja siinä määrin, että muutoksia voi olla mahdotonta kumota. Siksi yhä useampi tutkimus pyrkii tunnistamaan amyloidiplakkien olemassaolon jo aiemmassa vaiheessa. Näyttöä konventionaalisten, taudin oireita lievittävien lääkkeiden mahdollisimman varhaisen käytön hyödyistä on osoitettu myös suomalaisessa aineistossa (20).
Biomarkkerien ja kuvantamismenetelmien ja niihin liittyvien raja-arvojen käyttö on myös vaihdellut tutkimuksissa, koska täyttä selvyyttä normaalien arvojen määritelmästä ei toistaiseksi ole ollut. National Institute of Aging (NIA) ja sen Alzheimerin tautia tutkiva asiantuntijaryhmä on kehittänyt yhtenäiset ohjeet diagnostiikalle, tautivaiheen määrittelylle ja biomarkkerien käytölle erityisesti kliinisiä tutkimuksia ajatellen (18,21). Tämän uskotaan tulevaisuudessa yhdenmukaistavan ja tehostavan potilaiden valintaa ja diagnostiikkaa.
Oman haasteensa kehitykselle ovat asettaneet myös kognition muutosten havainnointiin käytettävät menetelmät. The Alzheimer’s disease Assessment Scale – Cognitive (ADAS-Cog) (22) aiheutti aluksi paljon variaatiota tulosten tulkinnassa. Menetelmän käyttökelpoisuutta kuvaavassa tutkimuksessa osoitettiin, että raportoijan kokemus vaikutti tulosten tulkintaan merkittävästi (3). ADAS-Cogin rinnalle onkin kehitetty uusi The Alzheimer’s Disease Composite Score (ADCOMS), joka perustuu erilaisiin yhdistettyihin pisteytyksiin (23). Nämä pisteytykset on kehitetty ADAS-Cogista, kliinisestä dementian asteen arvioinnista ja Mini-Mental State Examination -testistä (MMSE). MMSE on lyhyt kognitiivinen testisarja, joka kartoittaa aivojen toimintaa eri osa-alueilla: orientaatio, muisti, tarkkaavaisuus (laskemisen avulla tutkittuna), kieli (nimeäminen, toistaminen ja kirjoittaminen) sekä hahmotuskyky ja toiminnan ohjaus (kuvion kopiointi). ADCOMS on validoitu varhaisista kognitiivisista muutoksista kärsivien potilaiden tutkimuksissa, joissa osoitettiin sopiva pisteytyksen raja-arvo sellaisille potilaille, joita tarkastellessa kyettäisiin näkemään vaikutus lumelääkkeen ja tutkimuslääkkeen välillä yhden vuoden aikana. Menetelmä on osoitettu hyödylliseksi varhaisen vaiheen taudista kärsiville potilaille, joilla on hyvin vähän kognitiivisia muutoksia (23).
Lisäksi kliinisten tutkimusten reaktiivisten muutosten mahdollistamiseksi on kehitetty erilaisia malleja (22,24). Ns. bayesialaiset adaptiiviset tutkimusmallit mahdollistavat lääketutkimuksen jo alkuvaiheessa kerätyn tiedon hyödyntämisen loppututkimuksen aikana mm. lääkkeiden annostuksen, tutkimuksen keston ja potilasmäärän muokkaamiseen. Tämän avulla myös tutkimusten hyödyllisyyttä eli futiliteettia voidaan tarkastella ja ennustaa sekä selvittää, ovatko tutkimuksen ennalta asetetut tavoitteet vielä tutkimuksen aikana saavutettavissa. Silloin kannattamattomat tutkimukset voidaan lopettaa varhain tai muokata niitä niin, että niistä olisi eniten hyötyä myös tutkittaville potilaille.
Lopuksi
Tutkimuksissa, joissa amyloidiplakkien määrää potilaiden aivoissa on saatu vähennettyä, ei aina kuitenkaan ole nähty muutoksia kognition heikkenemisen etenemisen estämisessä (4,25). Toisaalta hiljattain on myös nähty tuloksia, joissa amyloidiplakkien määrän vähenemisen aivoissa on osoitettu voivan olla hyödyllinen ja jopa hidastavan kognitiivista heikkenemistä ainakin joissakin potilasryhmissä (26). Parhaillaan jännitetäänkin sitä, saako tutkimuksessa käytetty adukanumabi Yhdysvaltojen lääkeviranomaisen FDA:n hyväksyntää, ja jos saa, niin missä potilasryhmässä.
Myös elämäntapa- ja ravitsemusmuutoksilla on saatu lupaavia tuloksia erityisesti varhaisen tautivaiheen hoidossa (22,27). Monimuotoinen hoitokokonaisuus, joka sisältää elämäntapamuutoksia ja lääkehoitoa, voikin olla tulevaisuudessa keskeisessä asemassa.
Käsitys amyloidipeptidien roolista taudin kulussa on myös muuttunut. Tutkimusten perusteella amyloidioligomeerit voivat vaikuttaa neuronien toimintaan ja aiheuttaa synapsien toimintahäiriöitä monilla tavoilla, ja siksi odotukset taudin aiheuttamien vahinkojen korjaamisesta ovat muuttuneet. Lisäksi on opittu, että perustutkimusta metaboliasta, amyloidiplakkien monimuotoisuudesta, toksisuudesta ja molekyylirakenteista sekä yksilöllisistä immuunivasteista täytyy vielä merkittävästi täydentää.
Amyloidikuormaa vähentävä hoito pitäisi siis aloittaa ajoissa, mutta hoito ei toisaalta saisi olla liian tehokasta, jotta se ei häiritsisi amyloidiproteiinien luonnollisia fysiologisia tehtäviä. Amyloidihypoteesi ei siis ole kuollut, vaan se elää ja kehittyy edelleen. Toivoa menestyksekkäämmästä tulevaisuudesta ja mahdollisuudesta parantavampiin hoitoihin luovat viime vuosista huomattavasti monipuolistuneemmat uudet lääkevaikutusten kohteet sekä myös geeni- ja soluhoitojen tutkimus Alzheimerin taudissa.
Sauli Vuoti: Lääketieteellinen asiantuntija (Janssen-Cilag).
- 1
- Prince MJ. World Alzheimer report 2015: the global impact of dementia: an analysis of prevalence, incidence, cost and trends. London: Alzheimer’s disease international 2015.
- 2
- Rosenberg PB, Nowrangi MA, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer’s disease: What might be associated brain circuits? Mol Asp Med 2015;43–44:25–37.
- 3
- Mehta D, Jackson R, Pau G, Shi J, Sabbagh M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs 2017;26:735–9.
- 4
- Cummings J, Lee G, Ritter A, Sabbagh M. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement 2019;5:272–93.
- 5
- Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991;6:487–98.
- 6
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987;325:733–6.
- 7
- Gunawardena S, Yang G, Goldstein LS. Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum Mol Genet 2013;22:3828–43.
- 8
- Kolata G. Down syndrome--Alzheimer’s linked. Science (New York, NY). 1985;230:1152–3.
- 9
- Puzzo D, Gulisano W, Arancio O, Palmeri A. The keystone of Alzheimer pathogenesis might be sought in Abeta physiology. Neuroscience 2015;307:26–36.
- 10
- Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW. Beta-amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A 2018;115:4483–8.
- 11
- Kinney J, Bemiller M, Murshaw A, Leisgang A, Salazar A, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:575–90.
- 12
- Gomez-Nicola D, Boche D. Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimers Res Ther 2015;7:42.
- 13
- Zimmer ER, Leuzy A, Benedet AL, Breitner J, Gauthier S, Rosa-Neto P. Tracking neuroinflammation in Alzheimer’s disease: the role of positron emission tomography imaging. J Neuroinflammation 2014;11:120.
- 14
- Jack CR Jr, Knopman DS, Jagust WJ ym. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010;9:119–28.
- 15
- Gomez-Isla T, Hollister R, West H ym. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 1997;41:17–24.
- 16
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992;42(3 Pt 1):631–9.
- 17
- Bierer LM, Hof PR, Purohit DP ym. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol 1995;52:81–8.
- 18
- Jack CR Jr, Bennett DA, Blennow K ym. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dementia: J Alzheimer’s Assoc 2018;14:535–62.
- 19
- Dubois B. The emergence of a new conceptual framework for Alzheimer’s disease. J Alzheimers Dis 2018;62:1059–66.
- 20
- Linna M, Vuoti S, Silander K ym. Impact of anti-dementia medication on the risk of death and causes of death in Alzheimer’s disease. J Alzheimers Dis 2019;71:1297–308.
- 21
- Wang J, Logovinsky V, Hendrix SB, Stanworth SH, Perdomo C, Xu L. ADCOMS: a composite clinical outcome for prodromal Alzheimer’s disease trials. J Neurol Neurosurg Psychiatry 2016;87:993–9.
- 22
- Hendrix S, Soininen H, van Hees J ym. Alzheimer’s disease composite score: a post-hoc analysis using data from the LipiDiDiet trial in prodromal Alzheimer’s disease. J Prev Alzheimers Dis 2019;6:232–6.
- 23
- U.S. Food and Drug Administration. 2013. Guidance for Industry Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease. Food and Drug Administration, Washington, D.C. 2013.
- 24
- U.S. Food and Drug Administration, U.S. Department of Health and Human Services. Guidance for Industry; Early Alzheimer’s Disease: Developing Drugs for Treatment. Washington, DC: 2018.
- 25
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608.
- 26
- Schneider L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurology 2020;19:111–2.
- 27
- Soininen H, Solomon A, Jelle Visser P, Hendrix S, Blennow K, Kivipelto M. 24-month intervention with a specific multinutrient in people with prodromal Alzheimer’s disease (LipiDiDiet): a randomised, double-blind, controlled trial. Lancet Neurol 2017;16:965–75.
What is delaying the emergence of new Alzheimer medication?
Presently, the drugs available for treatment of Alzheimer’s Disease (AD), including cholinesterase inhibitors and antagonists of the N-methyl-D-aspartate receptor, can only inhibit dementia symptoms for a restricted period of time, but cannot stop or overturn disease progression. Based on the amyloid hypothesis, many global drug companies have conducted several clinical trials on amyloid clearing therapy without success. The number of anti-amyloid trials decreased in 2019, which might be a turning point and sign of a shift in focus in clinical trials. An in-depth and comprehensive understanding of the contribution of the amyloid hypothesis and other factors of AD is crucial for developing novel pharmacotherapies.
In ongoing clinical trials, researchers have developed and are currently testing several possible interventions aimed at various targets, including anti-amyloid and anti-tau interventions, neurotransmitter modification, anti-neuroinflammation and neuroprotection interventions, and cognitive enhancement and interventions to relieve behavioural psychological symptoms.







{kind=link}
{kind=link}