Perinnöllinen haimasyöpäalttius – kansallinen seurantasuositus
• Noin 10–15 % haimasyövistä johtuu perinnöllisestä alttiudesta.
• Mikäli haimasyöpäpotilaan lähisuvussa on todettu haimasyöpää tai hän on sairastunut alle 50-vuotiaana, suositellaan geneettisiä jatkotutkimuksia perinnöllisyyslääketieteen yksikössä.
• Suurimmassa osassa suvuista, joissa on perinnöllisyysselvityksissä todettu perinnöllinen haimasyöpäalttius, tarkka geneettinen etiologia jää epäselväksi.
• Seurannaksi suositellaan vuosittaista magneettikuvantamista pääsääntöisesti 50 ikävuodesta alkaen ja erittäin suuren riskin geenivirheissä endoskooppista kaikututkimusta joka toinen vuosi.
• Varhaisvaiheessa todetun haimasyövän viiden vuoden elossaololuku on noin 80 %.
Haiman duktaalinen adenokarsinooma on yleisin haiman pahanlaatuinen kasvain (1). Haimasyövän ilmaantuvuus on ollut viime vuosikymmeninä kasvussa erityisesti taloudellisesti kehittyneissä maissa. Muuttuva ikärakenne ja parantunut diagnostiikka selittävät osaltaan tätä ilmaantuvuuden kasvua, mutta myös ikävakioitu ilmaantuvuus on kasvanut (2).
Suomessa haimasyövän ilmaantuvuus on kasvanut, mutta ikävakioitu ilmaantuvuus on syöpärekisterin mukaan ollut suunnilleen samaa luokkaa 1980-luvulta lähtien (3).
Vuonna 2021 Suomessa todettiin 1 190 uutta haimasyöpätapausta. Riskin sairastua haimasyöpään 80 ikävuoteen mennessä on arvioitu olevan noin 1,4 % (3,4). Haimasyöpä on naisilla kolmanneksi ja miehillä neljänneksi yleisin syöpäkuolleisuutta aiheuttava syöpätauti Suomessa. Haimasyöpien ilmaantuvuus kasvaa 70 ikävuoden jälkeen, ja noin 65 % kaikista tapauksista Suomessa todetaan yli 70-vuotiaana (4).
Haimasyöpädiagnoosin viivästyminen on tavallista, sillä sappitietukoksen aiheuttamaa keltaisuutta lukuun ottamatta oireet ovat varsin epäspesifejä kuten ylävatsakipu, pahoinvointi ja laihtuminen. Jos haimasyöpä todetaan riittävän varhaisessa vaiheessa, potilaalle voidaan tarjota kirurgian ja solunsalpaajahoitojen yhdistelmiä, jotka parantavat ennustetta merkittävästi.
Tutkimuksissa on todettu, että leikatuista potilaista hieman yli 20 % on elossa viiden vuoden kohdalla (5,6). Mikäli haimasyöpä löydetään hyvin varhaisessa vaiheessa, viiden vuoden elossaololuku on jopa 84 % (7).
Riskitekijät
Haimasyövälle altistavia hyvin tunnettuja ulkoisia riskitekijöitä ovat tupakointi, diabetes, ylipaino ja krooninen haimatulehdus (2). Alkoholin suurkulutuksen on todettu olevan mahdollinen itsenäinen riskitekijä, mutta vähintäänkin se altistaa krooniselle haimatulehdukselle (2) (taulukko 1). Lisäksi tunnetaan haiman premaligneja muutoksia, joihin liittyy kohonnut haimasyöpäriski (8,9).

Haimasyövistä noin 10–15 %:n arvioidaan johtuvan perinnöllisestä alttiudesta (10,11,12,13,14,15,16). Perinnöllinen haimasyöpäalttius voi esiintyä osana tunnettua syöpäalttiusoireyhtymää, joka aiheuttaa merkittävästi lisääntyneen riskin sairastua erilaisiin syöpäsairauksiin taustalla olevasta ituradan geenivirheestä riippuen. Se voi myös liittyä perinnölliseen haimatulehdukseen (taulukko 2) tai esiintyä suvuittain ilman, että se liittyy tunnettuun syöpäalttiusoireyhtymään tai että sen taustalla on tunnistettu haimasyövälle altistavaa geenivirhettä (familial pancreatic cancer, FPC).
Tunnetuissa syöpäalttiusoireyhtymissä, perinnöllisissä haimatulehduksissa sekä suvuittain esiintyvässä haimasyöpäalttiudessa potilaan terveillä sukulaisilla on suurentunut riski sairastua haimasyöpään. Riskiä pidetään suurena, kun elämänaikainen haimasyöpäriski on yli 5 % tai suhteellinen riski on yli 5-kertainen (17). Taustalla olevasta geenivirheestä riippuen elämänaikainen riski on 3–53 % (taulukko 2).
Suuressa riskissä oleville terveille sukulaisille suositellaan seurantaa, koska se mahdollistaa syövän varhaisemman diagnostiikan.
Perinnöllisen haimasyöpäalttiuden työryhmä
Tämän kansallisen perinnöllistä haimasyöpäalttiutta koskevan seurantasuosituksen tavoite on yhtenäistää nykyisiä seurantakäytäntöjä ja vähentää haimasyöpäkuolleisuutta henkilöillä, joilla on perinnöllinen alttius sairastua.
Asetetussa työryhmässä ovat edustettuina kaikki Suomen yliopistosairaalat ja lääketieteen erikoisaloista ovat edustettuina perinnöllisyyslääketiede, vatsaelinkirurgia, gastroenterologia ja vatsaradiologia.
Seurantasuositukset pohjautuvat kansainvälisiin seurantasuosituksiin (18,19,20) huomioiden suomalaisen terveydenhuoltojärjestelmän rakenteet ja resurssit.
Perinnöllinen haimasyöpäalttius
Syöpäalttiusoireyhtymät
Haimasyövän riski on suurentunut erilaisissa perinnöllisissä syöpäalttiusoireyhtymissä. Syöpäalttiusoireyhtymiin liittyviä geenejä ovat STK11-, CDKN2A-, ATM-, TP53-, BRCA2- ja PALB2 -geenit (taulukko 2). Näiden geenien ituradan patogeenisten varianttien kantajilla on seurannan kannalta kliinisesti merkittävä riski sairastua haimasyöpään.
Nykyään tunnettuihin syöpäalttiusoireyhtymiin liittyy yleensä suurempi riski sairastua muihin syöpäkasvaimiin kuin haimasyöpään (10,12,13,16,21). Syöpäalttiusoireyhtymät esitellään tarkemmin kappaleessa Perinnölliset haimasyövälle altistavat geenivirheet.
Perinnöllinen haimatulehdus
Perinnölliseen haimatulehdukseen liittyy selkeästi kohonnut riski sairastua haimasyöpään. Siihen liittyvistä suuren riskin geeneistä yleisin on PRSS1 (13) (taulukko 2). Muita geenejä ovat muun muassa SPINK1 ja CFTR, mutta näihin geeneihin liittyvät haimasyöpäriskit ovat vähemmän tunnettuja (22,23).
Suvuittain esiintyvä haimasyöpäalttius
Suvuittain esiintyvä haimasyöpäalttius (FPC) ei liity tunnettuihin syöpäalttiusoireyhtymiin. Haimasyöpäsuvuissa voidaan todeta useassa sukupolvessa ja/tai usealla henkilöllä samassa sukupolvessa haimasyöpää, mutta perinnöllisen alttiuden geneettinen syy jää geenitutkimuksista huolimatta tuntemattomaksi (16).
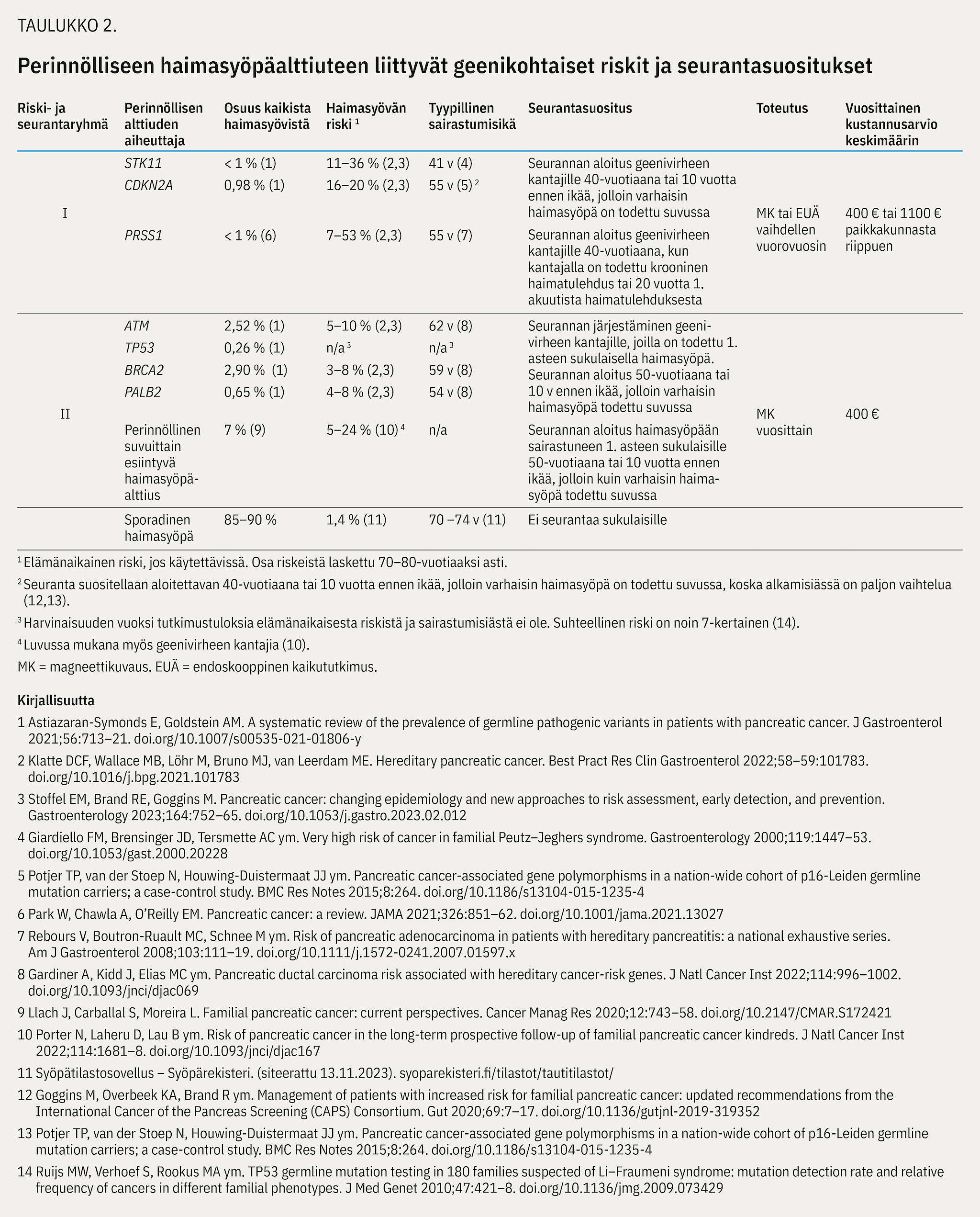
Suvuittain esiintyvän haimasyöpäalttiuden kriteerit täyttyvät, kun suvussa on vähintään kahdella keskenään 1. asteen sukulaisella todettu haiman duktaalinen adenokarsinooma (kuva 1) eikä geenitutkimuksissa ole todettu tunnettuihin syöpäalttiusoireyhtymiin liittyvää geenivirhettä (15). Haimasyövän riskin suuruus arvioidaan suvun sairaustietojen perusteella (taulukko 2).
Perinnöllisen haimasyöpäalttiuden riski vaihtelee
Jo yhdellä 1. asteen sukulaisella todettu sporadinen haimasyöpä kaksinkertaistaa henkilön riskin sairastua haimasyöpään (24). Perinnöllisessä haimasyöpäalttiudessa riski sairastua on tuoreen tutkimuksen mukaan 85 ikävuoteen mennessä noin 5 %, kun yhdellä 1.asteen sukulaisella on todettu haimasyöpä ja vastaavasti 9 % ja 18 %, kun kahdella tai kolmella 1. asteen sukulaisella on todettu haimasyöpä (25).
Kun yhdellä sukulaisella on todettu haimasyöpä alle 50-vuotiaana, riskiluvut ovat vastaavasti suuremmat: noin 7 %, 12 % ja 24 % (25). Haimasyöpäsuvuissa on kirjallisuudessa kuvattu myös antisipaatiota, eli haimasyöpä voi ilmetä seuraavissa sukupolvissa aikaisempaa nuoremmalla iällä (26).
Riskiä on vaikea arvioida suvuittain esiintyvässä haimasyöpäalttiudessa, kun geenitutkimuksessa ei todeta selittävää muutosta. Julkaistuihin sarjoihin sisältyy usein henkilöitä, joilla on todettu geenivirhe. Tuoreen tutkimuksen mukaan nuorimman haimasyöpäpotilaan sairastumisiän mediaani on näissä haimasyöpäsuvuissa 56 vuotta (27).
Perinnöllisiin syöpäalttiusoireyhtymiin ja perinnöllisiin haimatulehduksiin liitetty haimasyöpäriski vaihtelee eri tutkimuksissa. Geenivirheet voidaan jakaa kahteen ryhmään syöpäriskin ja seurantasuosituksen perusteella: erittäin suuri riski (I) ja suuri riski (II) (taulukko 2).
I) Erittäin suuren riskin potilasryhmään, jossa haimasyöpäriski on 7–53 %, kuuluvat kaikki STK11- ja CDKN2A- sekä tietyt PRSS1 -geenivirheiden kantajat (13,28). Tätä potilasryhmää suositellaan seurattavan tehostetusti riippumatta siitä, onko suvussa todettu haimasyöpää.
II) Suuren riskin potilasryhmään kuuluvat ATM-, TP53-, BRCA2- ja PALB2 -geenivirheiden kantajat, joilla haimasyöpäriski on noin 3–10 % (13,28). ATM-, TP53-, BRCA2- ja PALB2 -geenivirheiden kantajille suositellaan seurantaa, jos 1. asteen sukulaisella on haimasyöpä, koska nykykäsityksen mukaan kyseisillä geenivirheen kantajilla on suvussa suurin riski sairastua haimasyöpään.
BRCA2 -geeniin liittyy merkittävästi suurempi haimasyöpäriski kuin BRCA1 -geenivirheeseen (28,29,30,31). PALB2 -geeniin liittyvä elämänaikainen haimasyöpäriski on kohtalainen, jollei suvussa ole haimasyöpään sairastuneita henkilöitä. Pääosin riski on 2–3 %, mutta osalla henkilöistä riski vastaa BRCA2 -geenivirheen riskiä (28,32). Asiantuntija-arvioiden mukaan riski on todennäköisesti selvästi suurempi, jos potilaan 1. asteen sukulaisella on todettu haimasyöpä (13,30). PALB2 -geeniin liittyvään yleiseen suomalaiseen valtamutaatioon liittyvää haimasyöpäriskiä ei vielä tunneta riittävästi.
Nykyinen seurantasuositus on geenikohtainen, mutta tulevaisuudessa tutkimustiedon karttuessa suosituksia voidaan linjata myös varianttikohtaisesti.
Suuren riskin potilaisiin kuuluvat myös suvuittain esiintyvän haimasyöpäalttiuden kriteerit täyttävät suvut (18), ja heille suositellaan seurantaa.
BRCA1-, MLH1-, MSH2-, MSH6- ja EPCAM -geenivirheiden kantajilla on todettu kohtalainen riski sairastua haimasyöpään, mutta nykytiedon valossa riski ei eroa merkittävästi väestötason riskistä eikä edellytä seurantaa (13,32,33,34). Potilasryhmän kohdalla suositellaan harkitsemaan seurantaa sukuhistorian perusteella riskissä oleville henkilöille käyttäen riskiryhmän II seurantaohjelmaa.
PMS2 -geenin geenivirheet ovat harvinaisia, ja niihin liittyy todennäköisesti pienempi syöpäriski kuin muihin MMR-geeneihin (mismatch-repair), joten myöskään PMS2 -geeniä ei sisällytetä nykyiseen seurantasuositukseen (33,35).
Perinnölliseen haimatulehdukseen liittyvistä geeneistä ei PRSS1 :tä lukuun ottamatta ole vielä tarpeeksi tietoa seurannan tueksi (22,23).
Lähettämisperusteet perinnöllisyysselvittelyihin
Kansainvälisissä tutkimuksissa noin 10 %:lla haimasyöpäpotilaista on todettu jokin haimasyövälle altistava geenivirhe geenipaneelitutkimuksessa (11,12,36,37). National Comprehensive Cancer Network (NCCN) suosittelee geenitestausta kaikille haimasyöpäpotilaille (38).
Suomessa kaikki haimasyöpäpotilaat, joiden 1. tai 2. asteen sukulaisella on todettu haimasyöpä, suositellaan lähettämään perinnöllisyyslääketieteen poliklinikalle perinnöllisyysneuvontaa, sukuanamneesin tarkentamista ja geenitutkimusten harkintaa varten.
Lisäksi on suositeltavaa tehdä lähete poikkeuksellisen nuorella iällä (alle 50-vuotiaana) sairastuneista sekä niistä, joilla on aikaisemmin todettu johonkin syöpäalttiusoireyhtymään sopiva toinen syöpäkasvain, vaikka suvussa ei muutoin olisi todettu haimasyöpää.
Lähetteen voi tehdä myös suoraan perinnöllisyyslääketieteen poliklinikalle perusterveydenhuollosta tai yksityiseltä sektorilta potilaasta, jolla itsellään ei ole haimasyöpää, kun potilaan suvussa on todettu kaksi haimasyöpää keskenään 1. tai 2. asteen sukulaisilla (kuva 1, kuvio 1).

Diagnostista geenipaneelitutkimusta suvussa kulkevan geenivirheen etsimiseksi tehdään vain potilaille, joilla on todettu haimasyöpä. Kun suvussa on todettu haimasyöpäpotilaalla syöpäalttiusoireyhtymään liittyvä geenivirhe, terveille sukulaisille voidaan tarjota kattavaa perinnöllisyysneuvontaa. Sen jälkeen suvussa esiintyvän geenivirheen ennustava geenitutkimus on mahdollinen sukulaisille, jotka ovat sen voineet periä. Seurannasta voidaan vapauttaa ne sukulaiset, joilla ei todeta suvussa esiintyvää syöpäalttiusoireyhtymälle altistavaa geenivirhettä.
Terveiden sukulaisten ennustavat geenitutkimukset tehdään vain perinnöllisyyslääketieteen poliklinikan järjestäminä. Suuressa haimasyöpäriskissä olevat potilaat keskitetään yliopistosairaaloiden vatsaelinkirurgian poliklinikalle seurannan järjestämiseksi (kuvio 1).
Perinnölliset haimasyövälle altistavat geenivirheet
Perinnöllisen haimasyöpäalttiuden geenitutkimukset
Suomessa haimasyöpäpotilaille suositellaan tarjottavan geenipaneelitutkimus, missä tutkitaan vähintään STK11, CDKN2A, PRSS1, ATM, TP53, BRCA1, BRCA2, PALB2, MLH1, MSH2, MSH6, EPCAM ja PMS2. Geenipaneelien sisällöt päivittyvät laboratorioissa säännöllisesti uusien tutkimustulosten ja suositusten mukaan.
STK11
STK11 -geeni (OMIM 602216) on kasvunrajoitegeeni, joka osallistuu solusyklin säätelyyn. STK11 -geenivirheet aiheuttavat autosomaalisesti vallitsevasti periytyvän Peutz–Jeghersin oireyhtymän, joka on perinnöllinen suoliston polypoositauti. Oireyhtymään liittyvät mukokutaaniset pigmenttiläiskät ja suoliston hamartoomat sekä lisääntynyt riski sairastua useisiin eri syöpäkasvaimiin kuten haimasyöpään, suolistosyöpään, rintasyöpään ja gynekologisiin syöpäkasvaimiin (39).
CDKN2A
CDKN2A -geeni (OMIM 600160) on kasvunrajoitegeeni, joka osallistuu solusyklin säätelyyn. CDKN2A -geenivirheet aiheuttavat autosomaalisesti vallitsevasti periytyvän FAMMMPC-oireyhtymän (familial atypical multiple mole and melanoma-pancreatic syndrome tai melanoma-pancreatic syndrome). Oireyhtymään liittyy noin 70 %:n riski sairastua melanoomaan ja noin 20 %:n riski sairastua haimasyöpään (40).
PRSS1
PRSS1 -geenin (OMIM 276000) koodaama trypsinogeeni on haiman erittämän ruuansulatusentsyymin trypsiinin esiaste, jonka ennenaikainen aktivoituminen haimakudoksessa aiheuttaa haimatulehduksen. PRSS1 -geenin geenivirheet aiheuttavat autosomaalisesti vallitsevasti periytyvää perinnöllistä haimatulehdusta, johon voi liittyä toistuvia akuutteja haimatulehduksia ja taudin edetessä krooninen haimatulehdus (41).
ATM
ATM -geeni (OMIM 607585) on kasvunrajoitegeeni, joka osallistuu DNA:n vaurioiden korjaukseen. ATM -geenivirheet on liitetty autosomaalisesti vallitsevasti periytyvään perinnölliseen rintasyöpään ja haimasyöpään sekä autosomaalisesti peittyvästi periytyvään ataksia-telangiektasiasairauteen (42).
TP53
TP53 -geeni (OMIM 191170) voi toimia sekä kasvunrajoitegeeninä että onkogeeninä. Geenin toiminnan lopettavat variantit (loss-of-function) estävät kasvunrajoittamisen kasvaimissa. Geenissä esiintyy myös aminohappomuutoksen aiheuttavia gain-of-function-variantteja, jotka aikaansaavat uusia toimintoja, jotka johtavat kasvaimen syntyyn (43).
TP53 -geenivirheet aiheuttavat autosomaalisesti vallitsevasti periytyvää Li–Fraumenin oireyhtymää ja perinnöllistä TP53 -geeniin liittyvää syöpäalttiusoireyhtymää, joihin liittyy riski sairastua useisiin eri syöpäkasvaimiin kuten rintasyöpään, pehmytkudos- ja luusarkoomaan, aivokasvaimiin, lisämunuaiskuoren syöpään ja haimasyöpään (44).
BRCA1, BRCA2 ja PALB2
BRCA1- (OMIM 113705), BRCA2- (OMIM 600185) ja PALB2 -geenit (OMIM 610355) ovat kasvunrajoitegeenejä, jotka osallistuvat DNA:n vaurioiden korjaukseen homologisen rekombinaation mekanismilla (45,46). BRCA1-, BRCA2- ja PALB2 -geenivirheet periytyvät autosomaalisesti vallitsevasti, ja niihin liittyy haimasyöpäriskin lisäksi suuri riski sairastua rintasyöpään sekä geenikohtaisesti vaihteleva riski sairastua munasarjasyöpään (32,45).
MLH1, MSH2, MSH6, EPCAM ja PMS
MLH1- (OMIM 120436), MSH2- (OMIM 609309), MSH6- (OMIM 600678) ja PMS (OMIM 2600259) -geenit osallistuvat DNA:n kahdentumisvirheiden korjaukseen muodostamalla MMR-mekanismin. EPCAM -geeni (OMIM 185535) ei kuulu MMR-mekanismiin, mutta sen deleetiot aiheuttavat MSH2 -geenin hypermetylaation, joka sammuttaa MSH2 -geenin toiminnan (47).
MMR-geenivirheet aiheuttavat autosomaalisesti vallitsevasti periytyvää Lynchin oireyhtymää ja harvinaista lapsuusiässä alkavaa autosomaalisesti peittyvästi periytyvää CMMRD-oireyhtymää (constitutional MMR deficiency syndrome).
Lynchin oireyhtymään liittyy geenikohtaisesti vaihteleva riski sairastua erilaisiin syöpäkasvaimiin kuten paksu- ja peräsuolensyöpään, kohtusyöpään, mahasyöpään, munasarjasyöpään, virtsateiden syöpään ja haimasyöpään (12,33,35).
Perinnöllisen haimasyöpäalttiuden kansallinen seurantaohjelma
Seurannan tavoitteena on vähentää haimasyöpäkuolleisuutta ja parantaa ennustetta tunnistamalla haimasyöpä mahdollisimman varhaisessa vaiheessa.
Seurannan hyödyt ja kustannustehokkuus
Kansallinen seurantasuositus on luotu kansainvälisten seurantasuositusten pohjalta. Se on rajattu erittäin suuren ja suuren riskin seurantaryhmiin, koska näyttöä ei ole vielä kattavasti kohtalaisen riskin geenien ryhmässä.
Seurantaryhmiin sisällytetään vain yksilöt, joiden riskin sairastua haimasyöpään arvioidaan olevan tutkitusti selvästi kohonnut verrattuna väestön riskiin. Geenivirheen kantajien riski on todennäköisesti vielä taulukon 2 riskiä suurempi silloin, kun 1. asteen sukulaisella on todettu haimasyöpä. Tämä on huomioitu seurantakriteereissä.
Mitä vähäisemmän riskin henkilöitä seurataan, sitä enemmän löytyy sattumalöydöksiä, joihin kohdistuu kirurgisia toimenpiteitä ja niihin sekä itse seurantaan liittyviä haittoja. Seurannan tehon pysyminen riittävänä suhteessa siihen kohdistuvaan resurssiin edellyttää, että potilaalla on suuren riskin geenivirhe tai suvuittain esiintyvän haimasyöpäalttiuden kriteerit täyttyvät.
Seuranta tullaan toteuttamaan julkisessa terveydenhuollossa, minkä vuoksi kustannustehokkuuden arviointi on suuremmassa roolissa kuin kansainvälisissä rekisteritutkimuksissa yleensä on. Haimasyöpätyöryhmämme linja on se, että suuren riskin yksilöiden seuranta tulee mahdollistaa julkisen terveydenhuollon kautta, koska tällä hetkellä terveydenhuollossa ei ole vaihtoehtoista kustannuspaikkaa.
Seurantakäytännöt ovat vaihdelleet sairaalakohtaisesti Suomessa, ja suosituksen tarkoitus on yhtenäistää seurantakäytäntöjä. Seurannassa olevia potilaita arvioidaan olevan yliopistosairaaloissa tällä hetkellä yhteensä noin 100.
Lisäämällä tietoisuutta perinnöllisestä haimasyöpäalttiudesta löydämme todennäköisesti uusia suuren riskin potilaita seurantaan. Perinnölliselle alttiudelle altistavat geenivirheet ovat väestössä harvinaisia ja seurantakriteerit tiukat, joten uusien seurantaan kuuluvien suuren riskin henkilöiden määrä ei todennäköisesti kasva kovin suureksi vuositasolla.
Tämänhetkisen tiedon perusteella suuren riskin geenivirheiden kantajien seuranta on perusteltua ja tehokasta sekä mahdollistaa haimasyöpien toteamisen varhaisessa vaiheessa ja siten parantaa merkittävästi diagnoosin jälkeistä elossaoloaikaa (18,48,49,50,51,52).
Kansainvälisissä laajoissa meta-analyyseissä merkittävien päätetapahtumien NNS-luvut (number needed to screen) ovat olleet 111–135 (53,54). Päätetapahtumien kokonaisesiintyvyydeksi suuren riskin yksilöillä arvioidaan kansainvälisissä tutkimuksissa 36–48 kk:n mediaaniseuranta-ajalla noin 2,25–3,3 % (49,52).
Vuosittainen seuranta magneettikuvauksella (MK) on tällä päätetapahtumafrekvenssillä todettu kustannustehokkaaksi (ICER eli incremental cost–effectiveness ratio alle 50 000 € / laatupainotettu elinvuosi) sekä tanskalaisessa että amerikkalais-hollantilaisessa kustannusvaikuttavuusanalyysissä (55,56). Sen sijaan tuoreen amerikkalaisen mallinnuksen mukaan seuranta MK:n ja endoskooppisen kaikututkimuksen yhdistelmällä oli kustannustehokasta vain erittäin suuren riskin yksilöillä (RR > 12) (57).
Vastaavasti toinen amerikkalainen tutkimus tarkasteli vuosittaisen endoskooppisella kaikututkimuksen avulla toteutetun seurannan tuloksia. Siinä esitettiin, että seuranta on kustannustehokasta, jos haimasyövän riski on yli 10 %, jäljellä oleva elossaolo on yli 16 vuotta ja jos endoskooppisella kaikututkimuksella jää diagnosoimatta alle 5 % tapauksista (58).
Suomalaisessa väestössä haimasyövälle altistavien geenivirheiden ja päätetapahtumien määrää ei tunneta eikä NNS-arvoa voida siten määrittää. Perinnöllisen haimasyöpäalttiuden työryhmän on tarkoitus perustaa kansallinen perinnöllisen haimasyöpäalttiuden rekisteri, jolloin seurantaa halutessaan suuren riskin yksilöt osallistuvat prospektiiviseen seurantatutkimukseen.
Seurantatutkimuksesta saadaan ajan kuluessa luotettava arvio vaikuttavuudesta ja kustannuksista. Seurantaohjelmaa päivitetään säännöllisesti kansainvälisten suositusten, uuden tutkimustiedon ja asiantuntija-arvioiden mukaisesti.
Seurannan toteutus Suomessa
Seurannan aloitusikä ja seurannan toteutus ovat geenikohtaisia (taulukko 2). Suurimmassa osassa suvuista seuranta tulisi aloittaa 50-vuotiaana tai 10 vuotta aikaisemmin kuin varhaisin haimasyöpä on todettu suvussa. Poikkeuksia ovat erittäin suuren riskin STK11-, CDKN2A- ja PRSS1 -patogeenisten varianttien kantajat, joiden kohdalla seuranta tulisi aloittaa aiemmin.
Seurantaväliksi suositellaan 12 kuukautta, mikäli seurannassa ei todeta poikkeavia löydöksiä. Työryhmä suosittaa rutiininomaisen seurannan päättämistä 75-vuotiaana tai aikaisemmin, jos potilas ei sovellu haimakasvaimen kirurgiseen hoitoon. CA19-9-testiä ei suositella, ellei kuvantamisessa löydy poikkeavaa.
I) Erittäin suuren riskin geenien (STK11, CDKN2A, PRSS1) kohdalla suositellaan seurantaa kaikille geenivirheen kantajille ylävatsan magneettikuvauksella yhdistettynä magneettikolangiografiaan (MK/MRCP) tai endoskooppisella kaikututkimuksella vuosittain ja vaihdellen menetelmää vuorovuosina.
II) Suuren riskin geenien (ATM, TP53, BRCA2, PALB2) kohdalla suositellaan seurantaa geenivirheen kantajille käyttäen MK/MRCP:tä vuosittain, mikäli vähintään yhdellä 1. asteen sukulaisella on todettu haimasyöpä. Suuren riskin suvuittain esiintyvässä haimasyöpäalttiudessa suositellaan seurantaa MK/MRCP:tä käyttäen vuosittain.
Magneettikuvaus seurannassa
Seurantakuvantamisen tulee olla tarkkuudeltaan riittävää mahdollisten alkuvaiheen muutosten toteamiseksi, eikä se saa aiheuttaa seurattavalle merkittävää haittaa. Soveltuvin kuvantamismenetelmä haiman kuvantamisseurannassa on MK, sillä oikein ja laadukkaasti toteutettuna se on resoluutioltaan hyvä eikä altista ionisoivalle säteilylle.
Kuvaukseen kuuluvat sekä tehoste- että MRCP-sarjat. Tarvittaessa tehostetuista kuvista tehdään myös subtraktiosarjat, jotka helpottavat tehostumisen tarkastelua.
Suositeltu kuvausprotokolla sekä lausuntojen laatimisen ohjeistus on kirjattu liiteaineistoon (liite 1, taulukko 3). Vakiomuotoinen lausuntopohja on suositeltava, koska se yhtenäistää arviointia ja parantaa lausuntojen laatua. Tietokonekerroskuvaus (TT) haimatuumoriprotokollalla (59) on tarpeen jatkoselvittelynä silloin, jos magneettikuvassa todetaan epäilyttävää, potilaalla on vaikeatulkintainen krooninen haimatulehdus tai MK:lla ei saada syystä tai toisesta diagnostisia kuvia.

Endoskooppinen kaikututkimus seurannassa
Kansainvälisissä suosituksissa (CAPS, AGA, ASGE) suositellaan käyttämään seurannassa joko MK:ta tai endoskooppista kaikututkimusta tai näiden yhdistelmiä. Endoskooppisella kaikututkimuksella voidaan löytää pienet, kiinteät muutokset paremmin kuin MK:lla (27,60,61) tai TT:llä (62,63).
Endoskooppisella kaikututkimuksella voidaan ottaa myös näytteitä tutkimuksen aikana epäilyttävistä kohteista. Tyypillisiä varhaisen haimasyövän löydöksiä ovat niukkakaikuiset pesäkkeet, haimatiehyeen laajentuma tai stenoosi. Endoskooppisella kaikututkimuksella voidaan arvioida myös haimakystia (64).
Huolimatta kaikututkimuksen hyvistä puolista, tutkimus on kuitenkin tekijä- ja kokemusriippuvainen (65,66,67). Laadukas haimakudoksen arviointi vaatii riittävän määrän tutkimuksia vuodessa ja ajantasaisen laitteiston. Kuvien tarkastelu jälkikäteen edellyttää myös tutkimuksen tallennusmahdollisuutta.
Endoskooppisen kaikututkimuksen komplikaatioriski on vähäinen (70,71), mutta sen suorittaminen vaatii esilääkitystä, sedaatiota tai anestesiaa. Varsinkin anestesiassa tehtynä menetelmä on kalliimpi kuin MK.
Suomessa endoskooppisen kaikututkimuksen käyttökokemus on lisääntynyt viime vuosina. Työryhmä suosittaa menetelmän käyttöä erittäin suuren riskin potilasryhmälle vuorovuosin MK:n kanssa, jos paikallinen resurssitilanne sen sallii.
Kansainväliset suositukset eivät tue rutiininomaista profylaktista haimanpoistoa siihen liittyvän sairastuvuuden (diabetes, eksokriininen vajaatoiminta) ja kuolleisuuden (noin 2–5 %) vuoksi. Ainoastaan vaikeaan perinnölliseen haimatulehdukseen liittyvän PRSS1 -geenivirheen yhteydessä koko haiman poistoa voi harkita yksilöllisesti suuren syöpäriskin vuoksi.
Viime vuosina sekä diabeteksen että eksokriinisen vajaatoiminnan hoito koko haiman poiston jälkeen on huomattavasti parantanut. Lisäksi sen vaikutus elämänlaatuun on todettu olevan vähäinen (68).
Lopuksi
Kansallisen seurantasuosituksen tavoitteena on vähentää haimasyöpäkuolleisuutta ja yhtenäistää nykyisiä seurantakäytäntöjä eri hyvinvointialueilla. Tällä hetkellä ei tiedetä suomalaisessa väestössä esiintyvien suuren riskin geenivirheiden esiintyvyyttä haimasyöpäpotilailla tai seurantaan ohjautuvien potilaiden tarkkaa määrää.
Laskennallisesti Suomessa vuosittain todetusta noin 1 200 haimasyövästä noin 120–150 voisi selittyä perinnöllisellä haimasyöpäalttiudella. Näistä vain pienelle osalle on toistaiseksi tehty geenipaneelitutkimus.
Seurantasuosituksen tavoite on auttaa kliinikoita tunnistamaan perinnöllinen haimasyöpäalttius ja ohjata potilaat perinnöllisyysselvittelyihin. Tämän jälkeen seurannan organisointivastuu suositellaan keskittämään yliopistosairaaloiden vatsaelinkirurgian yksiköihin.
Työryhmän on tarkoitus päivittää seurantasuositusta säännöllisesti ja perustaa suomalainen perinnöllisen haimasyöpäalttiuden potilasrekisteri. Rekisterin avulla voimme arvioida seurannan toteutumista ja tehokkuutta.
Yksilöllinen geeniohjattu lääkehoito tulee ohjaamaan geenitutkimusten tarvetta haimasyöpäpotilailla. On todennäköistä, että tulevaisuudessa kaikille haimasyöpäpotilaille tarjotaan geenitutkimuksia jo diagnoosivaiheessa.
Kiitokset Suomen Perinnöllisyyslääkärit ry:lle, Suomen Gastroenterologiayhdistykselle, Suomen Gastrokirurgit ry:lle ja Suomen Vatsaradiologit yhdistykselle yhteistyöstä. Lisäksi kiitos Lynchin oireyhtymään perehtyneelle tenure track-professori ja ylilääkäri Toni Seppälälle kommentoinnista artikkeliin liittyen.
Sini Keskinen: Apurahat (Suomen Gastroenterologiayhdistys: kirjoitusapuraha, Tyks-säätiö), muu (väitöskirjatyö perinnöllisestä haimasyöpäalttiudesta Varhan alueella).
Reea Ahola: Apurahat (Suomen Gastroenterologiayhdistys: kirjoitusapuraha).
Kristiina Aittomäki: Ei sidonnaisuuksia.
Perttu Arkkila: Matka-, majoitus- tai kokouskulut (Mediq).
Maria Haanpää: Ei sidonnaisuuksia.
Outi Kuismin: Ei sidonnaisuuksia.
Pekka Lammi: Ei sidonnaisuuksia.
Johanna Louhimo: Ei sidonnaisuuksia.
Marika Mäkinen: Ei sidonnaisuuksia.
Minna Nortunen: Korvaus käsikirjoituksen kirjoittamisesta tai tarkistamisesta (Suomen Gastroenterologiayhdistys).
Minna Pöyhönen: Ei sidonnaisuuksia.
Irina Rinta-Kiikka: Apurahat (Suomen Gastroenterologiayhdistys: kirjoitusapuraha)
Minna Kankuri-Tammilehto: Luentopalkkiot (Pfizer: koulutustilaisuus Perinnöllinen syöpä – Moniammatillisia näkökulmia kliinikon arkeen).
Saila Kauhanen: Korvaus käsikirjoituksen kirjoittamisesta tai tarkistamisesta (Suomen Gastroenterologiyhdistys).
- 1
- Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J 2012;18:492–501. doi.org/10.1097/PPO.0b013e31827459b6
- 2
- Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol 2021;18:493–502. doi.org/10.1038/s41575-021-00457-x
- 3
- Seppä K, Tanskanen T, Heikkinen S, Malila N, Pitkäniemi J. Syöpä 2021. Tilastoraportti Suomen syöpätilanteesta. syoparekisteri.fi/raportit-ja-katsaukset/syopa-raportti/
- 4
- Syöpätilastosovellus – Syöpärekisteri. (siteerattu 13.11.2023). syoparekisteri.fi/tilastot/tautitilastot/
- 5
- Aaltonen P, Carpén O, Mustonen H ym. Long-term nationwide trends in the treatment of and outcomes among pancreatic cancer patients. Eur J Surg Oncol 2022;48:1087–92. doi.org/10.1016/j.ejso.2021.11.116
- 6
- Huang L, Jansen L, Balavarca Y ym. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: a large, international population-based study. BMC Med 2018;16:125. doi.org/10.1186/s12916-018-1120-9
- 7
- Blackford AL, Canto MI, Klein AP, Hruban RH, Goggins M. Recent trends in the incidence and survival of stage 1A pancreatic cancer: a surveillance, epidemiology, and end results analysis. J Natl Cancer Inst 2020;112:1162–9. doi.org/10.1093/jnci/djaa004
- 8
- European Study Group on Cystic Tumours of the Pancreas. European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018;67:789–804. doi.org/10.1136/gutjnl-2018-316027
- 9
- Ohtsuka T, Fernandez-Del Castillo C, Furukawa T ym. International evidence-based Kyoto guidelines for the management of intraductal papillary mucinous neoplasm of the pancreas. Pancreatology 2024;24:255–70. doi.org/10.1016/j.pan.2023.12.009
- 10
- Hu C, Hart SN, Polley EC ym. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 2018;319:2401–9. doi.org/10.1001/jama.2018.6228
- 11
- Gardiner A, Kidd J, Elias MC ym. Pancreatic ductal carcinoma risk associated with hereditary cancer-risk genes. J Natl Cancer Inst 2022;114:996–1002. doi.org/10.1093/jnci/djac069
- 12
- Kasuga A, Okamoto T, Udagawa S ym. Molecular features and clinical management of hereditary pancreatic cancer syndromes and familial pancreatic cancer. Int J Mol Sci 2022;23:1205. doi.org/10.3390/ijms23031205
- 13
- Klatte DCF, Wallace MB, Löhr M, Bruno MJ, van Leerdam ME. Hereditary pancreatic cancer. Best Pract Res Clin Gastroenterol 2022;58–59:101783. doi.org/10.1016/j.bpg.2021.101783
- 14
- Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: Advances and challenges. Cell 2023;186:1729–54. doi.org/10.1016/j.cell.2023.02.014
- 15
- Llach J, Carballal S, Moreira L. Familial pancreatic cancer: current perspectives. Cancer Manag Res 2020;12:743-758. https://doi.org/10.2147/CMAR.S172421
- 16
- Gentiluomo M, Canzian F, Nicolini A, Gemignani F, Landi S, Campa D. Germline genetic variability in pancreatic cancer risk and prognosis. Semin Cancer Biol 2022;79:105–131. doi.org/10.1016/j.semcancer.2020.08.003
- 17
- Canto MI, Harinck F, Hruban RH ym. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. doi.org/10.1136/gutjnl-2012-303108
- 18
- Goggins M, Overbeek KA, Brand R ym. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020;69:7–17. doi.org/10.1136/gutjnl-2019-319352
- 19
- Sawhney MS, Calderwood AH, Thosani NC ym. ASGE guideline on screening for pancreatic cancer in individuals with genetic susceptibility: summary and recommendations. Gastrointest Endosc 2022;95:817–26. doi.org/10.1016/j.gie.2021.12.001
- 20
- Aslanian HR, Lee JH, Canto MI. AGA clinical practice update on pancreas cancer screening in high-risk individuals: expert review. Gastroenterology 2020;159:358–62. doi.org/10.1053/j.gastro.2020.03.088
- 21
- Abe K, Kitago M, Kitagawa Y, Hirasawa A. Hereditary pancreatic cancer. Int J Clin Oncol 2021;26:1784–92. doi.org/10.1007/s10147-021-02015-6
- 22
- Le Cosquer G, Maulat C, Bournet B, Cordelier P, Buscail E, Buscail L. Pancreatic cancer in chronic pancreatitis: pathogenesis and diagnostic approach. Cancers Basel 2023;15:761. doi.org/10.3390/cancers15030761
- 23
- Greenhalf W, Lévy P, Gress T ym. International consensus guidelines on surveillance for pancreatic cancer in chronic pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and European Pancreatic Club. Pancreatology 2020;20:910–8. doi.org/10.1016/j.pan.2020.05.011
- 24
- Antwi SO, Fagan SE, Chaffee KG ym. Risk of different cancers among first-degree relatives of pancreatic cancer patients: influence of probands' susceptibility gene mutation status. J Natl Cancer Inst 2019;111:264–71. doi.org/10.1093/jnci/djx272
- 25
- Porter N, Laheru D, Lau B ym. Risk of pancreatic cancer in the long-term prospective follow-up of familial pancreatic cancer kindreds. J Natl Cancer Inst 2022;114:1681–8. doi.org/10.1093/jnci/djac167
- 26
- McFaul CD, Greenhalf W, Earl J ym. Anticipation in familial pancreatic cancer. Gut 2006;55:252–8. doi.org/10.1136/gut.2005.065045
- 27
- Overbeek KA, Levink IJM, Koopmann BDM ym. Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 2022;71:1152–60. doi.org/10.1136/gutjnl-2020-323611
- 28
- Stoffel EM, Brand RE, Goggins M. Pancreatic cancer: changing epidemiology and new approaches to risk assessment, early detection, and prevention. Gastroenterology 2023;164:752–65. doi.org/10.1053/j.gastro.2023.02.012
- 29
- Li S, Silvestri V, Leslie G ym. Cancer risks associated with BRCA1 and BRCA2 pathogenic variants. J Clin Oncol 2022;40:1529–41. doi.org/10.1200/JCO.21.02112
- 30
- Chen X, Meyer MA, Kemppainen JL ym. Risk of syndrome-associated cancers among first-degree relatives of patients with pancreatic ductal adenocarcinoma with pathogenic or likely pathogenic germline variants. JAMA Oncol 2023;9:955–61. doi.org/10.1001/jamaoncol.2023.0806
- 31
- Laish I, Schechter M, Dancour A ym. The benefit of pancreatic cancer surveillance in carriers of germline BRCA1/2 pathogenic variants. Cancer 2024;130:256–66. doi.org/10.1002/cncr.35052
- 32
- Yang X, Leslie G, Doroszuk A ym. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol 2020;38:674–85. doi.org/10.1200/JCO.19.01907
- 33
- Møller P, Seppälä TT, Bernstein I ym. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 2018;67:1306–16. doi.org/10.1136/gutjnl-2017-314057
- 34
- Dominguez-Valentin M, Haupt S, Seppälä TT ym. Mortality by age, gene and gender in carriers of pathogenic mismatch repair gene variants receiving surveillance for early cancer diagnosis and treatment: a report from the prospective Lynch syndrome database. EClinicalMedicine 2023;58:101909. doi.org/10.1016/j.eclinm.2023.101909
- 35
- Dominguez-Valentin M, Sampson JR, Seppälä TT ym. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 2020;22:15–25. doi.org/10.1038/s41436-019-0596-9
- 36
- Astiazaran-Symonds E, Goldstein AM. A systematic review of the prevalence of germline pathogenic variants in patients with pancreatic cancer. J Gastroenterol 2021;56:713–21. doi.org/10.1007/s00535-021-01806-y
- 37
- Rainone M, Singh I, Salo-Mullen EE, Stadler ZK, O'Reilly EM. An emerging paradigm for germline testing in pancreatic ductal adenocarcinoma and immediate implications for clinical practice: a review. JAMA Oncol 2020;6:764–71. doi.org/10.1001/jamaoncol.2019.5963
- 38
- Daly MB, Pal T, AlHilli Z ym. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 3.2024, The NCCN Clinical Practice Guidelines in Oncology, National Comprehensive Cancer Network. (siteerattu 20.5.2024). www.nccn.org/home/
- 39
- Beggs AD, Latchford AR, Vasen HF ym. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975–86. doi.org/10.1136/gut.2009.198499
- 40
- Overbeek KA, Rodríguez-Girondo MD, Wagner A ym. Genotype-phenotype correlations for pancreatic cancer risk in Dutch melanoma families with pathogenic CDKN2A variants. J Med Genet 2021;58:264–9. doi.org/10.1136/jmedgenet-2019-106562
- 41
- Whitcomb DC, Gorry MC, Preston RA ym. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141–5. doi.org/10.1038/ng1096-141
- 42
- Roberts NJ, Jiao Y, Yu J ym. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2012;2:41–6. doi.org/10.1158/2159-8290.CD-11-0194
- 43
- Hayashi A, Hong J, Iacobuzio-Donahue CA. The pancreatic cancer genome revisited. Nat Rev Gastroenterol Hepatol 2021;18:469–81. doi.org/10.1038/s41575-021-00463-z
- 44
- Guha T, Malkin D. Inherited TP53 mutations and the Li–Fraumeni syndrome. Cold Spring Harb Perspect Med 2017;7:a026187. doi.org/10.1101/cshperspect.a026187
- 45
- Zhao W, Wiese C, Kwon Y, Hromas R, Sung P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu Rev Biochem 2019;88:221–45. doi.org/10.1146/annurev-biochem-013118-111058
- 46
- Jones S, Hruban RH, Kamiyama M ym. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009;324:217. doi.org/10.1126/science.1171202
- 47
- Kuiper RP, Vissers LE, Venkatachalam R ym. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat 2011;32:407–14. doi.org/10.1002/humu.21446
- 48
- Klatte DCF, Boekestijn B, Onnekink AM ym. Surveillance for pancreatic cancer in high-risk individuals leads to improved outcomes: a propensity score-matched analysis. Gastroenterology 2023;164:1223–31.e4. doi.org/10.1053/j.gastro.2023.02.032
- 49
- Dbouk M, Katona BW, Brand RE ym. The multicenter cancer of pancreas screening study: impact on stage and survival. J Clin Oncol 2022;40:3257–66. doi.org/10.1200/JCO.22.00298
- 50
- Koopmann BDM, Omidvari AH, Lansdorp-Vogelaar I, Cahen DL, Bruno MJ, de Kok IMCM. The impact of pancreatic cancer screening on life expectancy: A systematic review of modeling studies. Int J Cancer 2023;152:1570–80. doi.org/10.1002/ijc.34379
- 51
- Paiella S, Capurso G, Carrara S ym. Outcomes of a 3-year prospective surveillance in individuals at high risk of pancreatic cancer. Am J Gastroenterol 2024;119:739–47. doi.org/10.14309/ajg.0000000000002546
- 52
- Signoretti M, Bruno MJ, Zerboni G, Poley JW, Delle Fave G, Capurso G. Results of surveillance in individuals at high-risk of pancreatic cancer: A systematic review and meta-analysis. United Eur Gastroenterol J 2018;6:489–99. doi.org/10.1177/2050640617752182
- 53
- Corral JE, Mareth KF, Riegert-Johnson DL, Das A, Wallace MB. Diagnostic yield from screening asymptomatic individuals at high risk for pancreatic cancer: a meta-analysis of cohort studies. Clin Gastroenterol Hepatol 2019;17:41–53. doi.org/10.1016/j.cgh.2018.04.065
- 54
- Kogekar N, Diaz KE, Weinberg AD, Lucas AL. Surveillance of high-risk individuals for pancreatic cancer with EUS and MRI: a meta-analysis. Pancreatology 2020;20:1739–46. doi.org/10.1016/j.pan.2020.10.025
- 55
- Joergensen MT, Gerdes AM, Sorensen J, Schaffalitzky de Muckadell O, Mortensen MB. Is screening for pancreatic cancer in high-risk groups cost-effective? – Experience from a Danish national screening program. Pancreatology 2016;16:584–92. doi.org/10.1016/j.pan.2016.03.013
- 56
- Corral JE, Das A, Bruno MJ, Wallace MB. Cost-effectiveness of pancreatic cancer surveillance in high-risk individuals: an economic analysis. Pancreas 2019;48:526–36. doi.org/10.1097/MPA.0000000000001268
- 57
- Peters MLB, Eckel A, Seguin CL ym. Cost-effectiveness analysis of screening for pancreatic cancer among high-risk populations. JCO Oncol Pract 2024;20:278–90. doi.org/10.1200/OP.23.00495
- 58
- Kumar S, Saumoy M, Oh A ym. Threshold analysis of the cost-effectiveness of endoscopic ultrasound in patients at high risk for pancreatic ductal adenocarcinoma. Pancreas 2021;50:807–14. doi.org/10.1097/MPA.0000000000001835
- 59
- Suomen Vatsaradiologit. Vatsan-TT-ohjeistus. (siteerattu 25.4.2024). view.officeapps.live.com/op/view.aspx?src=https%3A%2F%2Fsry.fi%2Fapp%2Fuploads%2Fsites%2F5%2F2021%2F06%2FVatsan-TT-ohjeistus.docx&wdOrigin=BROWSELINK
- 60
- Kitano M, Yoshida T, Itonaga M, Tamura T, Hatamaru K, Yamashita Y. Impact of endoscopic ultrasonography on diagnosis of pancreatic cancer. J Gastroenterol 2019;54:19–32. doi.org/10.1007/s00535-018-1519-2
- 61
- Harinck F, Konings IC, Kluijt I ym. A multicentre comparative prospective blinded analysis of EUS and MRI for screening of pancreatic cancer in high-risk individuals. Gut 2016;65:1505–13. doi.org/10.1136/gutjnl-2014-308008
- 62
- Ikemoto J, Serikawa M, Hanada K ym. Clinical analysis of early-stage pancreatic cancer and proposal for a new diagnostic algorithm: a multicenter observational study. Diagnostics Basel 2021;11:287. doi.org/10.3390/diagnostics11020287
- 63
- Krishna SG, Rao BB, Ugbarugba E ym. Diagnostic performance of endoscopic ultrasound for detection of pancreatic malignancy following an indeterminate multidetector CT scan: a systemic review and meta-analysis. Surg Endosc 2017;31:4558–67. doi.org/10.1007/s00464-017-5516-y
- 64
- Yamada R, Tsuboi J, Murashima Y, Tanaka T, Nose K, Nakagawa H. Advances in the early diagnosis of pancreatic ductal adenocarcinoma and premalignant pancreatic lesions. Biomedicines 2023;11:1687. doi.org/10.3390/biomedicines11061687
- 65
- Topazian M, Enders F, Kimmey M ym. Interobserver agreement for EUS findings in familial pancreatic-cancer kindreds. Gastrointest Endosc 2007;66:62–7. doi.org/10.1016/j.gie.2006.09.018
- 66
- Soares JB, Iglesias-Garcia J, Goncalves B ym. Interobserver agreement of EUS elastography in the evaluation of solid pancreatic lesions. Endosc Ultrasound 2015;4:244–9. doi.org/10.4103/2303-9027.163016
- 67
- Wani S, Han S, Simon V ym. Setting minimum standards for training in EUS and ERCP: results from a prospective multicenter study evaluating learning curves and competence among advanced endoscopy trainees. Gastrointest Endosc 2019;89:1160–8.e9. doi.org/10.1016/j.gie.2019.01.030
- 68
- Scholten L, Latenstein AE, Aalfs CM ym. Prophylactic total pancreatectomy in individuals at high risk of pancreatic ductal adenocarcinoma (PROPAN): systematic review and shared decision-making programme using decision tables. United Eur Gastroenterol J 2020;8:865–77. doi.org/10.1177/2050640620945534






