Tuberoosiskleroosi – suomalainen diagnoosi- ja seurantasuositus
•Tuberoosiskleroosia sairastavat potilaat tarvitsevat systemaattista, monen erikoisalan seurantaa läpi elämän.
•Tavoitteena on haitallisten tai jopa hengenvaarallisten elinmuutosten varhainen toteaminen ja hoito.
•Epilepsian tehokkaalla hoidolla ja kehityksen oikea-aikaisella tuella pyritään vaikuttamaan potilaiden kehitysennusteeseen ja neuropsykiatristen häiriöiden esiintymiseen.
•Tässä suosituksessa esitetään diagnosointi- ja seurantavaiheessa tarvittavat tutkimukset ja niiden toteutus, jossa hyödynnetään sekä erikoissairaanhoidon että perusterveydenhuollon palveluja.

Tuberoosiskleroosi (tuberous sclerosis complex, TSC) on monimuotoinen, useissa ektodermaalista alkuperää olevissa elimissä ilmenevä, vallitsevasti periytyvä neurokutaaninen harvinaissairaus. Sen ilmaantuvuus on noin 1/6 000 vastasyntynyttä ja esiintyvyys väestössä noin 1/20 000 (1). Sairaus todetaan osalla jo raskausaikana tai vastasyntyneenä, useimmilla lapsuusiässä, mutta joskus vasta aikuisena. Sairaus voi olla lähes oireeton tai aiheuttaa merkittäviä haittoja, kehityksen ongelmia ja hengenvaarallisia elinkomplikaatioita.
Tuberoosiskleroosia sairastavat tarvitsevat monialaista seurantaa koko ikänsä. Diagnosointia (taulukko 1) ja seurantaa varten on olemassa kansainvälinen suositus (1,2). Suomessa näiden potilaiden seurantaa ei ole keskitetty, kuten monissa Euroopan maissa. Meillä potilas saa hoidon lähellä kotiaan, mutta yksittäinen lääkäri kohtaa tuberoosiskleroosipotilaan vain harvoin. Tämän suosituksen tarkoituksena on helpottaa hoitavien lääkärien työtä ja yhtenäistää diagnostiikkaa ja seurantaa Suomessa (Liitetaulukko 1).
Ihomuutokset
Epäily tuberoosiskleroosista voi herätä pelkkien iho-oireiden perusteella, ja monet siihen liittyvät iholöydökset jäävät usein havaitsematta (3). Siksi taudin diagnosointivaiheessa suositellaan, että ihotautilääkäri tutkii potilaan, erityisesti jos diagnoosi on epävarma.
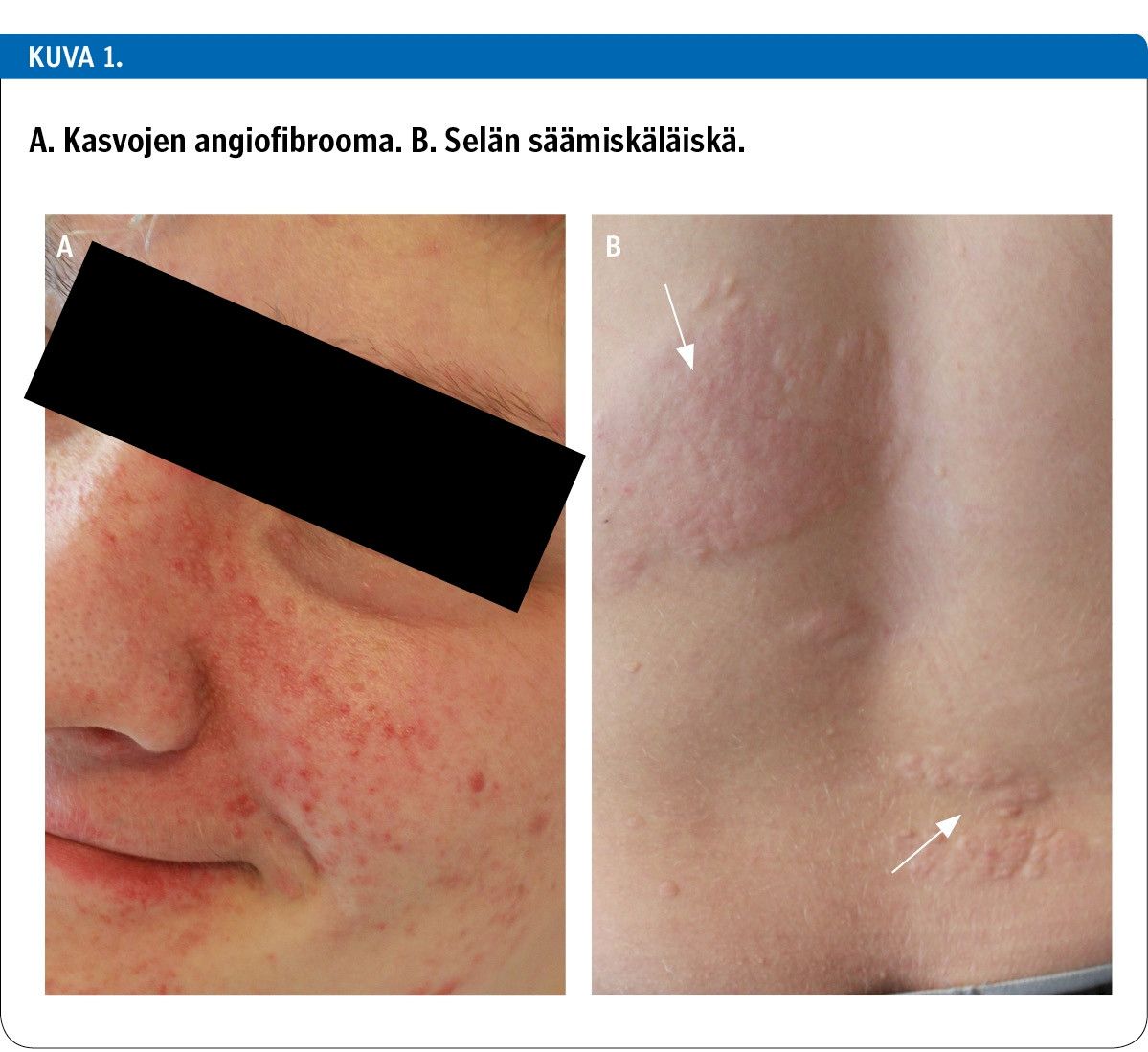
Tyypillisimpiä ihomuutoksia ovat saarnen lehden muotoiset hypopigmentoituneet läikät, kasvojen alueen papulaiset angiofibroomat, pään alueen fibroottiset plakit, kynnen alueen fibroomat, tavallisimmin alaselän alueelta löytyvät säämiskäläiskät sekä lukuisat pienet hypopigmentoituneet täplät (kuva 1). Useimpia näistä löydöksistä esiintyy yksittäisinä myös henkilöillä, joilla ei ole tuberoosiskleroosia (1). Hypopigmentoituneet läiskät voidaan havaita usein jo vastasyntyneellä, ja niiden diagnosoimisessa auttaa Woodin lamppu. Angiofibroomat ilmaantuvat tavallisesti 2–5 vuoden iässä, kynnen alueen fibroomat murrosiässä, ja säämiskäläiskät ennen kymmentä ikävuotta (4).
Taudin diagnosoinnin jälkeen iho-oireita on suositeltavaa seurata vuosittain (2). Seurannan voi toteuttaa potilaan hoitava lääkäri. Jos havaitaan ihomuutoksia, jotka vuotavat verta, kasvavat tai lisääntyvät nopeasti, aiheuttavat toiminnallista haittaa, ovat kivuliaita tai häiritsevät sosiaalista kanssakäymistä, potilas ohjataan ihotautilääkärille tarkempaa diagnosointia ja hoidon arviointia varten. Ihomuutoksia hoidetaan mm. laserilla, kirurgisesti sekä jäädytyshoidoin (4). Myös paikallisen mTOR:n estäjän tehosta kasvojen angiofibroomiin on julkaistu rohkaisevia tuloksia (5), mutta toistaiseksi kaupallista valmistetta ei ole saatavilla.
Neurologiset oireet
Epilepsia
Noin 80 % tuberoosiskleroosipotilaista sairastuu epilepsiaan ja noin 60 %:lla epilepsia on vaikea. Epilepsia puhkeaa 60 %:lle jo imeväisiässä paikallisalkuisena epilepsiana, infantiilispasmioireyhtymänä tai näiden yhdistelmänä. 10 %:lla epilepsia alkaa vasta aikuisiässä (6,7,8).
Imeväisiässä epilepsian viiveetön diagnosointi ja hoidon aloitus vaikuttavat lapsen kehitysennusteeseen (9). Myös vanhemmilla potilailla epilepsian hyvä hoito todennäköisesti tukee kognitiota.
Kun imeväisellä on todettu tuberoosiskleroosi, vanhempia opetetaan tunnistamaan epileptiset spasmit ja ensimmäisen ikävuoden aikana aivosähkökäyrää rekisteröidään toistuvasti. Näin varmistetaan hoidon varhainen aloittaminen. Lääkitys aloitetaan, kun EEG:ssä nähdään epileptisiin spasmeihin sopivia muutoksia tai paikallisia purkauksia, vaikka kliinisiä oireita ei vielä nähtäisikään (10,11). Imeväisiässä alkavan epilepsian ensisijainen lääkehoito on vigabatriini (10,12,13).
Kaikilta tuberoosiskleroosidiagnoosin saaneilta 1–16-vuotiailta lapsilta rekisteröidään uni-EEG. Jos herää epäily vähäoireisista kohtauksista, kuvataan pitkä video-EEG.
Tuberoosiskleroosiin liittyvän epilepsian lääkehoito määräytyy epilepsiatyypin mukaan. Vaikeaa epilepsiaa sairastavien hoitovaihtoehdot tulisi arvioida vaikean epilepsian hoitoon erikoistuneessa yksikössä. Lääkehoidon ohella potilaat voivat hyötyä epilepsiakirurgiasta, mTOR:n estäjästä, ruokavaliohoidosta tai stimulaattorihoidosta (14,15,16,17).
Jättisolukasvaimet
Aivojen toistuvan kuvantamisen tärkein päämäärä on havaita varhaisessa vaiheessa jättisolukasvaimet (subependymal giant cell astrocytoma, SEGA), joita esiintyy 10–20 %:lla tuberoosiskleroosipotilaista (18). Diagnoosivaiheessa suositellaan pään magneettikuvausta, jossa samalla nähdään taudin mahdolliset muut aivolöydökset kuten, tuberit ja ependyyminalaiset nodulukset (2). Jättisolukasvaimen tyypillinen esiintymispaikka on sivukammion kaudotalaaminen uurre lähellä aivokammioiden väliaukkoa (foramen Monroi).
Histologialtaan jättisolukasvaimet ovat hyvänlaatuisia, mutta kasvaessaan ne voivat aiheuttaa hydrokefaluksen. Ne voivat kasvaa myös invasiivisesti ympäröiviin aivorakenteisiin. Aivopaineoireiden lisäksi voi esiintyä paikallisia neurologisia tai neuropsykiatrisia oireita tai epilepsiakohtauksia.
Uusia jättisolukasvaimia ilmaantuu harvoin enää 25 ikävuoden jälkeen mutta olemassa oleva jättisolukasvain voi kasvaa vielä aikuisiässä (19,20). Kaikkien potilaiden pään magneettikuvausta 1–3 vuoden välein suositellaan 25 ikävuoteen asti (2,19,21). Harvajaksoista kuvantamisseurantaa läpi elämän suositellaan potilaille, joilla on aiemmin todettu jättisolukasvain.
Jättisolukasvain pyritään ensisijaisesti poistamaan kokonaan. Mikäli tämä ei ole mahdollista tai kirurgiselle toimenpiteelle on vasta-aiheita (esim. anestesia vasta-aiheinen), hoitovaihtoehtona on lääkehoito mTOR:n estäjillä (11,22, 23). Neurokirurgin konsultaatio tarvitaan aina, mikäli jokin kirurgisen hoidon aihe täyttyy tai kokonaistilanne on epäselvä (Liitetaulukko 1).
Käytös ja kehitys
Tuberoosiskleroosiin liittyy yleisesti kognitiivisen kehityksen häiriöitä, käytöshäiriöitä ja psyykkisiä häiriöitä, jotka ovat merkityksellisiä potilaan ja hänen perheensä elämänlaadulle. Kognitiivisen kehityksen eriasteisia häiriöitä esiintyy jopa puolella potilaista. Sen lisäksi oireyhtymään liittyvät neuropsykiatriset oireet (TSC-associated neuropsychiatric disorder, TAND) ovat yleisiä, mutta niitä ei aina tunnisteta (24). Kehityksellisten ja käyttäytymisongelmien tunnistamiseen ja seurantaan on kehitetty ns. TAND-kysely (25).
Diagnoosivaiheessa lapsipotilaan kehitystaso ja käyttäytymisen piirteet tulee kartoittaa lastenneurologian yksikössä. Työvälineeksi suositellaan TAND-kyselyä ja tarvittaessa moniammatillisen työryhmän tutkimusta laaja-alaisten kehityksen ongelmien diagnosoimiseksi, vanhempien ohjaamiseksi ja seurannan suunnittelemiseksi. Aikuisena diagnosoitujen potilaiden tutkimuksissa tulee käydä läpi koulutus- ja työhistoria sekä psyykkisten oireiden esiintyminen ja tarvittaessa ohjata lisäselvittelyihin. TAND-kysely sopii myös aikuispotilaille.
Lasten säännöllinen kehitysseuranta mahdollistaa oikea-aikaisen intervention. Vuosittaisilla seurantakäynneillä tulee seuloa kyselylomakkeella TAND-oireita, ja keskeisissä ikävaiheissa tulee suorittaa tarkempi, tarvittaessa moniammatillinen tutkimus. Suositellut tutkimusajankohdat ovat 0–3 vuotta, 3–6 vuotta, alakouluikä, teini-ikä ja varhainen aikuisikä, jonka jälkeen kognitiivista profiilia ja käytösoireita kartoitetaan tarpeen mukaan (2). Palvelu-, kuntoutus- ja pedagogisen suunnitelman tulee perustua hyviin hoito- ja kuntoutuskäytäntöihin (2). Käyttäytymisen tai kehitystason äkillinen muutos on hälytysmerkki perustaudin etenemisestä (epilepsia, jättisolukasvain, munuaissairaus) ja sen tulee johtaa tutkimuksiin.
Lasten kehitysseurannan ja kuntoutuksen suunnittelun vastuu on alueellisen työnjaon ja kehityksessä ilmenneiden ongelmien mukaisesti lastenneurologian poliklinikoilla tai kehitysvammapoliklinikoilla. Käytöshäiriöiden hoidossa ja seurannassa tehdään yhteistyötä lasten-, nuoriso- tai kehitysvammapsykiatrian kanssa. Käytösoireiden lääkehoidossa noudatetaan yksilöllistettyjä, mutta näyttöön perustuvia periaatteita (24).
Aikuisikäisten potilaiden TAND-oireiden seuranta toteutetaan ongelmien laadun mukaan kehitysvammapoliklinikalla, neurologian poliklinikalla tai omalääkärin vastaanotolla tarvittaessa psykiatrian klinikkaa konsultoiden.
Munuaiset
Tuberoosiskleroosiin liittyy munuaismuutoksia, ja ne lisääntyvät iän karttuessa: munuaislöydöksiä todetaan 2-vuotiaista noin 20 %:lla ja aikuisista jopa 80–90 %:lla.
Tavallisin munuaismuutos ovat yleensä multippeleina ja molemminpuolisina esiintyvät angiomyolipoomat, jotka ovat kasvutavaltaan hyvänlaatuisia ja yleensä oireettomia. Ne koostuvat rasvan lisäksi sileästä lihaksesta ja verisuonista. Niihin liittyy usein aneurysmia, jotka voivat altistaa spontaaneille, munuaistoimintaa heikentäville ja jopa hengenvaarallisille verenvuodoille (26). Verenvuotojen riskitekijöitä ovat kasvaimen suuri koko (yli 4 cm), nopea kasvutapa ja aneurysmien koko (yli 0,5 cm) (27).
Munuaiskystia esiintyy 30–45 %:lla potilaista (26). Kystien koko vaihtelee mikroskooppisen pienistä multippeihin, laajoihin muutoksiin. Vaikea ja varhainen polykystisen munuaistaudin (PKD) muoto on tavallisesti aiheutunut vierekkäisten kromosomissa 16p13-sijaitsevien TSC2- ja PKD1-geenien deleetiosta. Tuberoosiskleroosipotilaista 2–5 % sairastaa tätä tautimuotoa, ja heille munuaisten vajaatoiminta voi ilmaantua murrosiässä.
Harvinaisempia munuaismuutoksia ovat munuaiskarsinooma (aikuisista 3–5 %:lla) ja munuaiskivet.
Munuaisten vajaatoiminta etenee loppuvaiheeseen useimmiten siksi, että toimiva munuaiskudos vähenee muutosten kasvaessa, mutta myös verenvuotojen seurauksena. Tarpeettomat toimenpiteet tai lääkitykset edistävät vajaatoiminnan kehittymistä. Munuaisongelmat ovat aikuisten tuberoosiskleroosipotilaiden tavallisin kuolemansyy.
Munuaismuutosten ensisijainen kuvantamismenetelmä on magneettikuvaus. Koepalan ottamista tulee harkita, mikäli kuvantaen todettu muutos ei ole tyypillinen angiomyolipooma tai kysta. Pahanlaatuisuuteen viittaa kasvaimen nopea kasvutapa (yli 0,5 cm vuodessa), kalkkeutuminen tai nekroottisuus muutoksen keskellä (28). Munuaismuutoksia tulee seurata kuvantaen 1–3 vuoden välein ensisijaisesti magneettikuvauksin, mutta myös kaiku- tai TT-kuvausta voidaan käyttää. Munuaistoiminta määritetään diagnoosivaiheessa ja vähintään vuosittain verikokein ja tarvittaessa munuaiskerässuodoksen eli GFR-määrityksellä (2).
Viimeaikaisten tutkimusten perusteella mTOR:n estäjä vaikuttaa turvalliselta ja tehokkaalta hoidolta, jolla voidaan ehkäistä angiomyolipoomien kasvua, pienentää jo muodostuneita muutoksia ja säilyttää munuaistoimintaa. Hoito voidaan suunnitella aloitettavaksi jo varhaisessa vaiheessa ja vaikeissa tapauksissa aloittaa jo lapsuusiässä (29,30). Mikäli hoito lopetetaan, angiomyolipoomat kasvavat uudestaan, joten aloitettua hoitoa jatketaan koko eliniän.
Elektiivinen radiologinen selektiivinen angioembolisaatio (SAE) on oireettoman angiomyolipooman ensisijainen toimenpidehoitomuoto, mutta hoitoja voidaan joutua uusimaan ja munuaisen toiminta voi toimenpiteiden seurauksena heiketä (31). Elektiivinen munuaisen osapoisto on mielekäs vain sellaisissa harvoissa tilanteissa, joissa angiomyolipooma on yksittäinen ja pinnallinen (32). Vuotaneen, kipuja tai toistuvaa verivirtsaisuutta aiheuttavan angiomyolipooman ensisijainen hoito on angioembolisaatio (33).
Suu ja hampaat
Tuberoosiskleroosipotilaiden suuongelmien diagnosointi, ehkäisy ja hoito kannattaa keskittää. Yliopistosairaalan erikoishammaslääkärin on syytä arvioida tilanne mielellään jo diagnoosivaiheessa. Tutkimuksessa kiinnitetään huomiota sekä suun yleiseen terveydentilaan että sairauden tyypillisiin ilmentymiin suussa.
Yleisiä tuberoosiskleroosin aiheuttamia hammas- ja suumuutoksia ovat kiilteen kehityshäiriöt (kiilteen kuoppaisuus) erityisesti pysyvissä hampaissa. Muita ilmentymiä ovat suun sisäiset fibroomat, tavallisimmin ikenen alueella (34,35). Osalla potilaista on myös suuria haasteita suun päivittäisessä omahoidossa, ja heillä vaarana ovat hampaiden reikiintyminen ja kiinnityskudossairaudet.
Erikoishammaslääkäri arvioi potilaskohtaisesti tarvittavan seurannan tiheyden (tutkimusväli 6–12 kk), radiologisten tutkimusten tarpeellisuuden (ortopantomografia) ja seurantapaikan. Lapsipotilaiden tutkimuksesta ja hoitosuunnitelmasta vastaa lasten hammashoidon erikoishammaslääkäri. Potilaille tehdään henkilökohtainen hoitosuunnitelma yleisten suusairauksien ehkäisyyn ja hoitoon, ja siihen voi liittyä hammaslääkärin käyntejä sekä perusterveydenhuollossa että erikoissairaanhoidossa ja suuhygienistin ehkäisevää hoitoa. Suun fibroomia voidaan poistaa kirurgisesti esimerkiksi laserilla (4,36). Tätä tulee harkita erityisesti, jos liikakasvu haittaa hampaiden normaalia suuhun puhkeamista tai suun omahoitoa. Suun alueen fibroomien, luisten epämuodostumien ja puhkeamishäiriöiden hoito järjestetään suusairauksien yksikössä erikoisalojen yhteistyönä.
Sydänmuutokset
Tuberoosiskleroosipotilaista 50 %:lla on sydämen rabdomyoomia, ja toisaalta 59–80 %:lla rabdomyoomapotilaista todetaan tuberoosiskleroosi (37). Rabdomyoomat ovat hamartoomiksi luokiteltuja sydämen kasvaimia, ja 90 %:lla potilaista niitä on useita. Ne suurenevat raskauden aikana ja pienenevät syntymän jälkeen. Oireet ratkaisee kasvainten sijainti ja koko. 16 %:lla potilaista on merkittäviä rytmihäiriöitä (38).
Multippelit tai yksittäiset kookkaat rabdomyoomat havaitaan yleensä raskaudenaikaisissa seulontakaikukuvauksissa, ja silloin sikiökardiologin konsultaatio on aiheellinen. Lapsuusikäisille potilaille tehdään EKG-rekisteröinti ja sydämen kaikukuvaus. Jos epäillään rabdomyoomia, konsultoidaan lastenkardiologia. Lisätutkimuksina käytetään mm. EKG:n pitkäaikaisrekisteröintiä ja sydämen magneettikuvausta. Mikäli tuberoosiskleroosi todetaan aikuisiässä, johtumishäiriöiden ja arytmioiden diagnosoimiseksi tehdään EKG-tutkimus ja tarvittaessa sydämen kaikukuvaus.
Mikäli sikiöaikana todetaan arytmioita, pleura- tai perikardiumeffuusioita tai rabdomyoomat aiheuttavat verenkierron virtausesteen, ultraääniseuranta ja synnytys on syytä keskittää HYKS:aan, jossa on mahdollisuus välittömiin postnataalisiin sydänkirurgisiin toimenpiteisiin tai lääkehoidon harkintaan (mTOR:n estäjät).
Oireettomien potilaiden rabdomyoomia seurataan tiiviisti ensimmäisen elinvuoden ajan yksilöllisellä aikataululla lastenkardiologin tutkimuksin. Sen jälkeen seurantaväli on 1–3 vuotta. Lastenkardiologi arvioi seurantatarpeen potilaan siirtyessä aikuispuolelle. Kaikille potilaille suositellaan EKG-seurantaa 5 vuoden välein.
Keuhkomuutokset
Tyypillisin tuberoosiskleroosin keuhkoilmentymä on lymfangioleiomyomatoosi (LAM), jossa keuhkokudoksessa todetaan kystamuutoksia. Sitä esiintyy jopa 80 %:lla naispotilaista ja 10–12 %:lla miehistä.
Lymfangioleiomyomatoosin toteamiseksi suositellaan keuhkojen ohutleiketietokonetomografiaa (HRTT) kaikille, myös oireettomille, 18 vuotta täyttäneille naispuolisille ja oireisille miespuolisille tuberoosiskleroosipotilaille. Keuhkotoiminnan arvioimiseksi suositellaan tehtäväksi keuhkojen toimintakokeet ja 6 minuutin kävelytesti (6MWT). Seerumin vaskulaarisen endoteelikasvutekijän (VEGF-D) määritys voi helpottaa jatkossa lymfangioleiomyomatoosin kehittymisen ja etenemisen arvioinnissa.
Jokaisella seurantakäynneillä tulee arvioida, esiintyykö hengenahdistus- tai hengästymisoiretta, ja ohjeistaa tupakoimattomuuteen ja estrogeenin käytöstä pidättäytymiseen. HRTT uusitaan 5–10 vuoden välein oireettomille, joilla ei ole todettu lymfangioleiomyomatoosiin viittaavia löydöksiä, ja 2–3 vuoden välein niille, joilla on jo todettu lymfangioleiomyomatoosi. Keuhkojen toimintakokeet ja 6MWT uusitaan vähintään vuosittain oireisille ja jos HRTT:ssä on jo todettu lymfangioleiomyomatoosi.
mTOR:n estäjillä voidaan pyrkiä vakauttamaan keuhkotoimintaa ja parantamaan toimintakykyä, kun lymfangioleiomyomatoosi on kohtalainen tai vaikea tai se etenee nopeasti. Myös keuhkonsiirto on mahdollinen hoitovaihtoehto.
Silmämuutokset
Noin 50 %:lla tuberoosiskleroosipotilaista on verkkokalvon hamartoomia joko toisessa tai molemmissa silmissä (39,40). Yleensä hamartoomat ovat synnynnäisiä, täysin oireettomia, ja seurannassakin ne muuttuvat vain vähän. Ne eivät huononna näöntarkkuutta eivätkä vaadi aktiivista hoitoa.
Silmätutkimuksessa voidaan todeta myös silmäluomien angiofibroomia ja verkkokalvon pigmenttimuutoksia sekä hyvin harvoin mm. karsastusta, pseudokolobooma (silmän linssissä ja värikalvolla) ja värikalvon sektoraalista hypopigmentaatiota.
Silmätutkimus on aiheellinen diagnoosivaiheessa potilaan iästä riippumatta. Mikäli verkkokalvon hamartoomia todetaan, seuranta suositellaan järjestettäväksi 6–12 kuukauden välein 0–6 vuoden iässä ja noin joka toinen vuosi 7–16 vuoden iässä. Aikuisille riittävät harvemmat kontrollit.
Silmälääkäri arvioi seurannan tarpeen yksilöllisesti. Mikäli ensimmäisessä silmätutkimuksessa ei todeta löydöksiä, uusi tutkimus on aiheellinen, jos näkemisessä tai silmissä havaitaan muutoksia. Mikäli epilepsiaan on käytössä vigabatriinilääkitys, silmäseurannan tarve ja laajuus määräytyy vigabatriinin käytön ja potilaan iän mukaan.
Perinnöllisyysneuvonta
Äidit, joiden sikiöillä on todettu rabdomyoomia, lähetetään lähimmän yliopistosairaalan sikiötutkimusyksikköön tai äitiyspoliklinikalle.
Tuberoosiskleroosia epäiltäessä tehdään TSC1- ja TSC2-geenitutkimus, jonka pyytää joko hoitava lääkäri tai yliopistosairaalan kliinisen genetiikan tai perinnöllisyyslääketieteen yksikkö. Valtaosalla tutkimus varmistaa diagnoosin, ja yli 70 % mutaatioista on uusia (41). Perinnöllisyyslääketieteen yksikössä selvitetään suvun tilanne ja arvioidaan uusiutumisriski. Lapsipotilas perheineen lähetetään yksikköön uudelleen, jos perhesuunnittelu tulee ajankohtaiseksi tai jos geenitutkimus jää normaaliksi.
Lapsena tuberoosiskleroosin diagnoosin saaneet lähetetään perinnöllisyysneuvontaan, kun he ovat täysikäisiä. Lisäksi lähetetään pariskunnat, joissa sukuhistorian perusteella on tuberoosiskleroosin riski ja joissa perhesuunnittelu on ajankohtainen.
mTOR:n estäjät
mTOR:n estäjät sirolimuusi ja everolimuusi vähentävät täsmälääkkeenä tuberoosiskleroosin aiheuttavasta geenimutaatiosta johtuvaa mTOR-signaalikaskadin hyperaktiivisuutta. Niiden on osoitettu estävän jättisolukasvainten, munuaisten angiomyolipoomien ja keuhkojen lymfangioleiomyomatoosimuutosten kasvua (22,30) ja vähentävän paikallisalkuisia kohtauksia tuberoosiskleroosista johtuvassa epilepsiassa (15,42). Tutkimuksia tehdään myös vaikutuksista joihinkin muihin ilmentymiin. Toistaiseksi everolimuusi on rajoitetusti peruskorvattava jättisolukasvaimiin, joita ei voida leikata, ja aikuisten angiomyolipoomien hoitoon (Liitetaulukko 2).
Seurannan järjestäminen Suomessa
Tämä seurantasuositus noudattelee kansainvälistä suositusta. Kaikki näkökulmat sisältävän seurannan tulee jatkua läpi elämän. Myös potilaiden itsensä tulee tuntea seurantaohjelma. Harvinaissairausrekisterit varmistavat osaltaan tulevaisuudessa potilaiden seurannan toteutumisen.
Ehdotamme, että vaikean epilepsian hoitovaihtoehtojen arviot, sydämen rabdomyoomien ja lapsipotilaiden munuaismuutosten seuranta, jättisolukasvainten hoito, mTOR:n estäjähoidon toteutus, hammas-, suu- ja silmämuutosten diagnosointi ja seurannan ohjelmointi sekä perinnöllisyysneuvonta keskitetään yliopistosairaaloihin tai alueellisen työnjaon mukaisesti keskussairaaloihin yliopistosairaaloiden konsultaatiotuella. Muuten seurannassa hyödynnetään kattavaa terveydenhuoltojärjestelmäämme.
Lapsuusiässä seurannasta vastaavat ensisijaisesti lastenneurologit. Aikuisiän kynnyksellä jokaiselle potilaalle tulee sopia seurannan vastuuyksikkö, joka konsultoi tarvittaessa muita erikoisaloja. Seurannasta vastaava yksikkö valitaan potilaan keskeisten elinongelmien perusteella: se voi olla neurologian tai sisätautien poliklinikka, kehitysvammapoliklinikka tai erityisneuvola tai perusterveydenhuollon omalääkäri.
Liisa Metsähonkala: Epilepsialiiton varapj, luentopalkkio (Novartis), matka-, majoitus- tai kokouskulut (Novartis), advisory board (Eisai), asiantuntijaryhmän jäsen (Fennomedical, UCB).
Reetta Kälviäinen: Konsultointipalkkiot (Eisai, GW Pharmaceuticals, Orion, Sage, Sandoz, UCB), luentopalkkiot (Eisai, Orion, UCB), korvaus koulutusaineiston tuottamisesta (Sandoz), Epilepsialiiton asiantuntijatyöryhmä.
Harry Nisen: Kongressimatkakulut (Novartis).
Päivi Vieira: Luentopalkkiot (Orion, UCB), matka-, majoitus- tai kokouskulut (Genzyme, Novartis, Shire).
Heikki Alapulli, Maria Arvio, Anita Hiippala, Ulla Hodgson, Arto Immonen, Kristiina Kananen, Marjo Karvonen, Janne Kataja, Jussi Leppävirta, Tuire Lähdesmäki, Minna Pöyhönen, Ritva Vanninen, Kristiina Vasara: Ei sidonnaisuuksia.
- 1
- Northrup H, Darcy A, Krueger. International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243–54.
- 2
- Krueger DA, Northrup H . International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;44:255–65.
- 3
- Staley BA ym. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics 2011;127(1):e117–25.
- 4
- Teng JM ym. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol 2014;150:1095–101.
- 5
- Balestri R ym. Analysis of current data on the use of topical rapamycin in the treatment of facial angiofibromas in tuberous sclerosis complex. J Eur Acad Dermatol Venereol 2015;29:14–20.
- 6
- Vignoli A ym. Epilepsy in TSC: certain etiology does not mean certain prognosis. Epilepsia 2013;54:2134–42.
- 7
- Chu-Shore CJ ym. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–41.
- 8
- Curatolo P ym. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol 2015;22:259–73.
- 9
- Cusmai R ym. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav 2011;22:735–9.
- 10
- Curatolo P ym; TSC Consensus Meeting for SEGA and Epilepsy Management. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2012;16:582–6.
- 11
- Józwiak, Mandera M. Natural history and current treatment options for subependymal giant cell astrocytoma in tuberous sclerosis complex. Semin Pediatr Neurol 2015;22:274–81.
- 12
- Wheless JW ym. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord 2007;9:353–412.
- 13
- Pellock JM ym. Infantile spasms: a U.S. consensus report. Epilepsia 2010;51:2175–89.
- 14
- Zhang K ym. Predictors of seizure freedom after surgical management of tuberous sclerosis complex: a systematic review and meta-analysis. Epilepsy Res 2013;105:377–83.
- 15
- French JA ym. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:2153–63.
- 16
- Kossoff EH ym. Tuberous sclerosis complex and the ketogenic diet. Epilepsia 2005;46:1684–6.
- 17
- Moavero R ym. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst 2010;26:1495–504.
- 18
- Adriaensen ME ym. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 2009;16:691–6.
- 19
- Roth J ym. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol 2013;49:439–44.
- 20
- Tsai JD ym. Association between the growth rate of subependymal giant cell astrocytoma and age in patients with tuberous sclerosis complex. Childs Nerv Syst 2016;32:89–95.
- 21
- Nabbout R ym. Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psychiatry 1999;66:370–5.
- 22
- Franz DN ym. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013;381:125–32.
- 23
- Habib SL ym. Is mtor inhibitor good enough for treatment all tumors in TSC patients? J Cancer 2016;7:1621–31.
- 24
- Curatolo P ym. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol 2015;14:733–45.
- 25
- de Vries PJym. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015;52:25–35.
- 26
- De Waele L ym. Tuberous sclerosis complex: the past and the future. Pediatr Nephrol 2015;30:1771–80.
- 27
- Yamakado K ym. Renal angiomyolipoma: relationships between tumor size, aneurysm formation, and rupture. Radiology 2002;225:78–82.
- 28
- Rouvière O ym. Kidney damage due to tuberous sclerosis complex: management recommendations. Diagn Interv Imaging 2013;94:225–37.
- 29
- Pirson Y. Tuberous sclerosis complex-associated kidney angiomyolipoma: from contemplation to action. Nephrol Dial Transplant 2013;28:1680–5.
- 30
- Franz DN ym. Long-term use of everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One 2016;11:e0158476.
- 31
- Sooriakumaran P ym. Angiomyolipomata: challenges, solutions, and future prospects based on over 100 cases treated. BJU Int 2010;105:101–6.
- 32
- Boorjian SA ym. The role of partial nephrectomy for the management of sporadic renal angiomyolipoma. Urology 2007;70:1064–8.
- 33
- Ramon J ym. Renal angiomyolipoma: long-term results following selective arterial embolization. Eur Urol 2009;55:1155–61.
- 34
- Sparling JD ym. Oral findings in 58 adults with tuberous sclerosis complex. J Am Acad Dermatol 2007;56:786–90.
- 35
- Ammari MM ym. Oral findings in a family with tuberous sclerosis complex. Spec Care Dentist 2015;35:261–5.
- 36
- Eisen DB, Fazel N. Treatment of gingival fibromas using CO2 laser and electrosurgery in a patient with tuberous sclerosis. Dermatol Online J 2008;14:7.
- 37
- Hinton RB ym. Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the international tuberous sclerosis consensus group. J Am Heart Ass 2014;3:e001493.
- 38
- Miayke CY ym. Cardiac tumors and associated arrhythmias in pediatric patients, with observations on surgical therapy for ventricular tachycardia. J Am Coll Cardiol 2011;58:1903–9.
- 39
- Hodgson N ym. Ophthalmic manifestations of tuberous sclerosis: a review. Clin Exp Ophthalmol 2016;22. doi:10.1111/ceo.12806.
- 40
- Rowley SA ym. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol 2001;85:420–3.
- 41
- Nellist M ym. Targeted Next Generations Sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med Genet 2015;16:10.
- 42
- Krueger DA ym. Long-term treatment of epilepsy in tuberous sclerosis. Neurology 2016;87:2408–15.
- 43
- Peterson DE ym. Oral mucous injury caused by mammalian target of rapamycin inhibitors: emerging perspective on pathobiology and impact on clinical practise. Cancer Med 2016;5:1897–907.
- 44
- Sasongo TH ym. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst Rev 2016;7:CD011272.
Recommended guidelines for the surveillance of patients with tuberous sclerosis in Finland
Tuberous sclerosis is a rare genetic multiorgan disorder with considerable individual variation. The patients need systematic follow-up by several specialists throughout their lives in order to prevent harmful and potentially fatal complications and to optimize the patients’ early cognitive development and quality of life. In this paper, the authors present a proposal on the organization of diagnostics and follow-up of these patients in Finland. This proposal is based on the international recommendations but takes the local facilities and the special features of the Finnish health care system into account.




{kind=link}