FH-lasten hoidon tavoitteet täyttyvät useimmiten
Lähtökohdat Familiaalinen hyperkolesterolemia (FH) johtaa hoitamattomana valtimonkovettumistautiin. Lasten hoidon toteutumisesta ja tuloksista Suomessa on niukasti tietoa.
Menetelmät Selvitimme Kysin lastenklinikan hoidossa olleiden lasten FH-diagnoosit vuosina 2000–2020. Taustatietojen lisäksi tallensimme todetut geenivirheet, lipidiarvot ja toteutuneen lääkehoidon.
Tulokset Varmistettuja FH-diagnooseja löytyi yhteensä 45 (tyttöjä 21). Lasten ikä diagnosointihetkellä oli keskimäärin 7,5 vuotta (1,9–15,1 v). Todetuista LDL-reseptorigeenin mutaatioista tavallisimmat olivat FH-Pohjois-Karjala (50 %), FH-Helsinki (14 %) ja FH-Pogosta (7 %). Joka neljännen potilaan mutaatio ei kuulunut suomalaisiin valtamutaatioihin. Yleisimmin aloitettu statiinilääkitys oli pravastatiini (76 %), mutta hoitovastuun siirtyessä aikuisyksikköön käytössä oli yleisimmin atorvastatiini (60 %). Lääkehoitoa saaneiden potilaiden seerumin LDL-kolesterolin keskiarvo lääkehoidon alkaessa oli 5,4 mmol/l ja hoitovastuun siirtyessä 3,0 mmol/l (vähenemä 43 %).
Päätelmät Valtamutaatiotutkimus ei löydä kaikkia FH-lapsipotilaita. Hoitotavoitteeseen päästään valtaosalla potilaista. Pravastatiinin asemaa lasten FH:n ensisijaislääkkeenä tulee pohtia kriittisesti.
Familiaalinen hyperkolesterolemia (FH) aiheuttaa hoitamattomana varhaista valtimonkovettumistautia ja ennenaikaisia kuolemia (1,2). Plasman kolesteroli- ja LDL-kolesterolipitoisuudet suurenevat pian syntymän jälkeen, ja jo lapsuusiässä on todettavissa alkavia valtimomuutoksia (3,4).
LDL-reseptorien viallisuus tai puutos johtaa LDL-kolesterolipitoisuuden kasvuun verenkierrossa. Suomessa on todettu seitsemän LDL-reseptorigeenin (LDLR) valtamutaatiota: FH-Helsinki, -Pohjois-Karjala, -Turku, -Pori, -Pogosta, -11 ja -12. Ne kattavat yhteensä noin 80 % FH-tapauksista (3). FH:n voi aiheuttaa myös mutaatio APOB- tai PCSK9-geeneissä.
Heterotsygooteilla FH-potilailla plasman LDL-kolesterolipitoisuus on yleensä 2–3-kertainen terveiden henkilöiden pitoisuuksiin verrattuna. Hyvin harvinaisessa homotsygoottisessa tai yhdistelmäheterotsygoottisessa FH-taudissa pitoisuus voi kasvaa jopa 10-kertaiseksi (3,5).
Epäily lapsen FH-taudista herää sukuanamneesin, todetun dyslipidemian (ennen kaikkea suurentuneen LDL-kolesterolipitoisuuden) ja vain harvoin kliinisen kuvan (jänneksantoomat) perusteella. FH-diagnoosia pidetään varmana, jos alle 16-vuotiaalla lapsella tai nuorella plasman kolesterolipitoisuus on toistuvasti yli 6,7 mmol/l, LDL-kolesterolipitoisuus on yli 4,0 mmol/l, ja lisäksi lapsella tai hänen ensimmäisen tai toisen asteen sukulaisellaan on jänneksantoomia tai lapsella on osoitettu LDLR-, APOB-100- tai PCSK9-geenin mutaatio (3).
Hoidon tavoitteena on pienentää plasman LDL-kolesterolipitoisuutta. Käypä hoito -suosituksen mukainen LDL-kolesterolipitoisuuden tavoite FH-lapsella on alle 3,5 mmol/l, mutta valtimoterveyden kannalta ihanteellinen taso on alle 2,5 mmol/l (3).
Ruokavaliohoito aloitetaan heti kun dyslipidemia on todettu, ja se sisältää pehmeiden rasvojen suosimisen, eläinperäisen rasvan välttämisen ja kasvistanoli- tai -sterolituotelisän käyttöönoton. Ruokavaliohoidolla LDL-kolesterolipitoisuus laskee 5–20 % (3,6,7). Lisäksi huomiota kiinnitetään fyysiseen aktiivisuuteen ja tupakoinnin välttämiseen. Nämä eivät kuitenkaan yksin yleensä riitä laskemaan plasman LDL-kolesterolipitoisuutta tavoitetasolle.
Varhain aloitettu statiinihoito parantaakin sairauden ennustetta (8), mutta tutkimusten otoskoot ovat olleet pieniä ja niiden kesto on ollut yleensä lyhyt (9). Pisimmässä, 20 vuoden seurantatutkimuksessa lapsena aloitettu statiinilääkitys hidasti kaulavaltimon intima median paksuntumista ja ehkäisi sydän- ja verisuonisairauksia aikuisuudessa (10). Käypä hoito -suosituksen mukaan statiinilääkitys voidaan aloittaa 8–10 ikävuodesta alkaen, yksilöllisesti harkiten jo aiemminkin (3). Statiinilääkitys ei aiheuta lapsipotilaille merkittäviä haittavaikutuksia mm. kasvuun tai kehitykseen lumeryhmään verrattuna (11).
Statiinihoito aloitetaan pienimmällä suositellulla vuorokausiannoksella, joka on valmistekohtainen. Eri statiineilla on myös erilainen maksimivaikutus LDL-kolesterolin pienenemiseen: esimerkiksi rosuvastatiini on tehokkaampi kuin pravastatiini (3). Annosta voidaan suurentaa tai valmiste vaihtaa tehokkaampaan, kunnes hoitotavoite saavutetaan.
Muita lääkkeitä, joita voidaan käyttää ainoina lääkkeinä tai yhdistettynä statiineihin ovat etsetimibi ja PCSK9:n estäjät. Etsetimibi pienentää LDL-kolesterolipitoisuutta yli 20 %, eikä merkittäviä haittavaikutuksia ole raportoitu, mutta tutkimusnäyttöä lapsista on vähän (3).
Tämän tutkimuksen tavoitteena oli kartoittaa Kuopion yliopistollisen sairaalan lastentautien poliklinikassa vuosina 2000–2020 diagnosoitujen alle 16-vuotiaiden FH-potilaiden hoitoa ja hoitotuloksia.
Aineisto ja menetelmät
Kysin lastentautien poliklinikalla FH-diagnoosin 2000–2020 saaneet alle 16-vuotiaat lapset poimittiin Uranus-potilastietojärjestelmästä. Heidän potilaskertomuksistaan kerättiin tiedot FH-epäilyn aiheista, sukutaustasta, iästä, sukupuolesta, muista diagnooseista ja kasvusta.
Diagnoosin tekohetkellä lapsista oli ylipainoisia 18 % ja lihavia 9 % ja seurannan päättyessä vastaavat osuudet olivat 12 % ja 12 % (12). Plasman lipidiarvoihin mahdollisesti vaikuttavia muita diagnooseja löytyi neljältä lapselta: kaksi hypotyreoosia ja kaksi tyypin 1 diabetesta.
Löydetyistä 45 FH-potilaasta 21 (47 %) oli tyttöjä. Lopulliseen tarkasteluun otettiin mukaan 44 potilasta (kuvio 1).
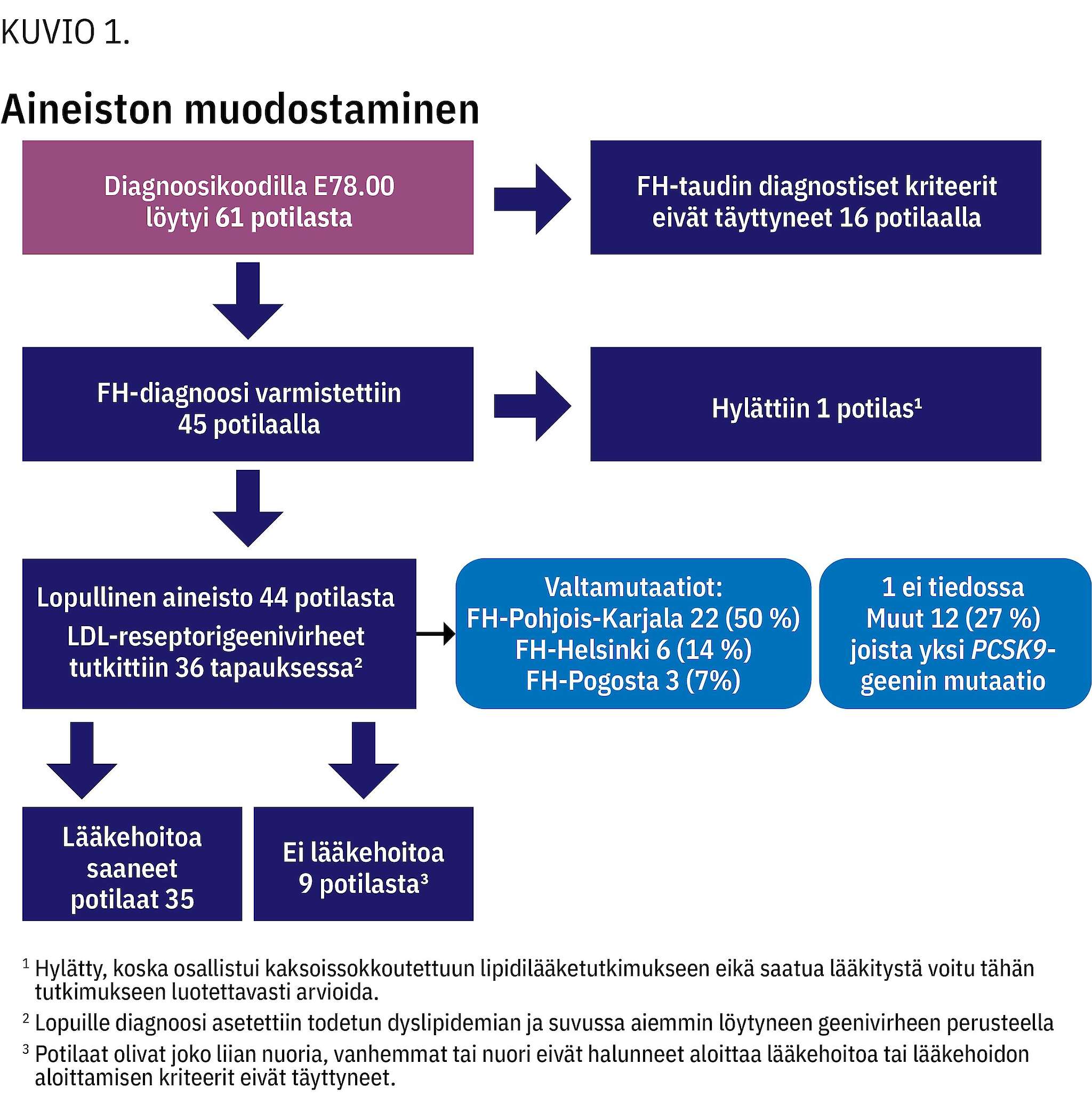
Diagnosointi-iän keskiarvo oli 7,5 v (vaihteluväli 1,9–15,1 v). FH-tautia lähdettiin tutkimaan suvussa todetun dyslipidemian vuoksi 30 tapauksessa (68 %) ja lapsella itsellään todetun dyslipidemian vuoksi 14 tapauksessa (32 %). Ksantoomia ei lapsilla todettu.
Potilaan tai ensimmäisen asteen sukulaisten FH-geenivirheet rekisteröitiin. LDL-reseptorigeenitutkimus oli tehty 36 lapselle. Muille diagnoosi asetettiin korkean kokonais- tai LDL-kolesterolitason ja sukuanamneesin perusteella. Lapsen tai suvun LDL-reseptorigeenin mutaatioista yleisimmät olivat FH-Pohjois-Karjala 22 (50 %), FH-Helsinki 6 (14 %) ja FH-Pogosta 3 (7 %). Muita mutaatioita todettiin sekvensoimalla 12 potilaalla (27 %). Yhdellä potilaalla oli PCSK9-geenin mutaatio. Yhdelle potilaalle diagnoosi oli asetettu ilman geenitestiä ja tietoa suvun tarkasta FH-tyypistä. Suvussa todettuja geenimutaatioita oli tiedossa yhteensä 38 potilaalla (86 %). Varhaisia valtimotapahtumia lähisukulaisilla oli 36 potilaalla (82 %).
Aineistosta selvitettiin myös toteutuneet hoidot, hoitovaste ja haittavaikutukset. Paikallisen hoito-ohjeen mukaan hoidossa tavoiteltiin LDL-kolesterolipitoisuutta alle 3 mmol/l. Jos tätä ei saavutettu, harkittiin statiiniannoksen suurentamista tai valmisteen vaihtoa tehokkaammaksi. Lipidiarvoista (kokonais-, LDL-, HDL- kolesteroli sekä triglyseridit) laskettiin keskiarvot ja 95 %:n luottamusvälit ennen lääkehoidon aloitusta, lääkehoidon aikana ja hoitovastuun siirtyessä aikuisyksikköön. Seurannan edelleen jatkuessa huomioitiin viimeiset tutkitut arvot. Muita hoitoja (elintapahoito, kasvistanolit tai -sterolit) ei tuloksissa erityisesti huomioitu, koska ruokavalio- ja muu elintapaohjaus oli annettu kaikille perheille heti diagnoosin varmistuttua.
Toissijaisesti arvioimme myös sairaanhoitopiirissä diagnosoitujen FH-lapsipotilaiden määrää suhteessa alueella seuranta-aikana syntyneiden lasten määrään.
Tilastolliseen tietojen käsittelyyn käytettiin SPSS 27.0.1-ohjelmaa (IBM SPSS Statistics for Windows, Version 27.0. Armonk, NY). Tutkimukselle oli saatu Kysin tutkimus- ja tietolupa.
Tulokset
Keskimääräinen kokonaisseuranta-aika oli 6,6 vuotta (vaihteluväli 0,9–11,6 v) ja lääkehoidon seuranta-aika 4,3 vuotta (0,1–11,5 v).
Diagnosointihetkellä kokonaiskolesterolin keskiarvo oli 7,4 mmol/l (vaihteluväli 4,8–9,9 mmol/l) ja LDL-kolesterolin 5,5 mmol/l (2,7–8,1 mmol/l).
Lääkehoito aloitettiin 35 potilaalle (80 %). Lääkehoidon aloitusiän keskiarvo oli 10,2 vuotta (4,1–15,5 vuotta). Yhdeksälle potilaalle ei seuranta-ajan päättyessä ollut aloitettu lääkehoitoa. Kahdessa tapauksessa perhe kieltäytyi lääkityksestä, muut lapset olivat niin nuoria, ettei lääkehoitoa vielä suositeltu.
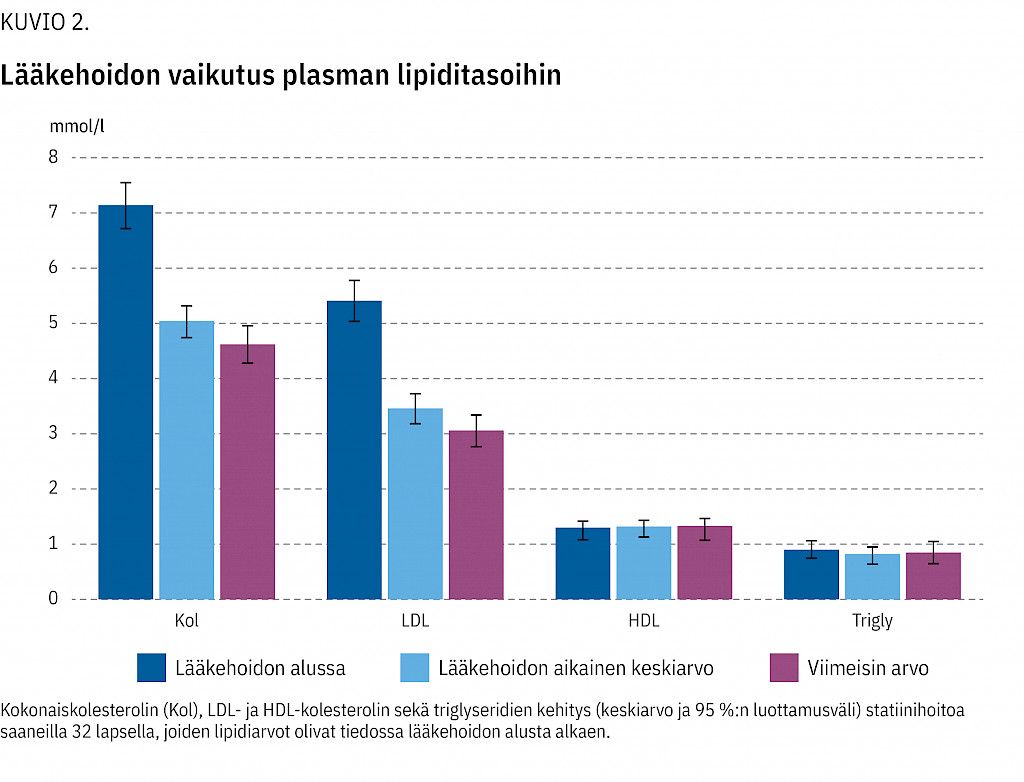
Lääkehoidon aikana (kaikki tarvittavat tiedot löydettiin 32 potilaalta) kolesterolipitoisuus pieneni keskimäärin 35 % (+6,3 % – –59,5 %) ja LDL-kolesterolipitoisuus 43 % (+6,7 % – –62,7 %) (kuvio 2). Lääkehoitoa saaneista 21 (60 %) potilasta pääsi seuranta-aikana hoitotavoitteeseen (LDL-kolesteroli ≤ 3,0 mmol/l).
Yhdelletoista potilaalle (31 %) kokeiltiin vain yhtä statiinivalmistetta ja heistä kahdeksan pääsi hoitotavoitteeseen. Yhdeksäntoista potilaan (54 %) lääkevalmiste vaihdettiin kerran ja viidellä (14 %) edettiin kolmanteen valmisteeseen. Toisella valmisteella tavoitteeseen pääsi 12 potilasta (34 %) ja kolmannella yksi potilas (3 %).
Lääkehoidon komplianssi arvioitiin hyväksi 27 potilaalla (77 %) ja vaihtelevaksi 8 potilaalla (23 %). Kukaan ei lopettanut lääkehoitoa kokonaan tai kieltäytynyt seurannasta.
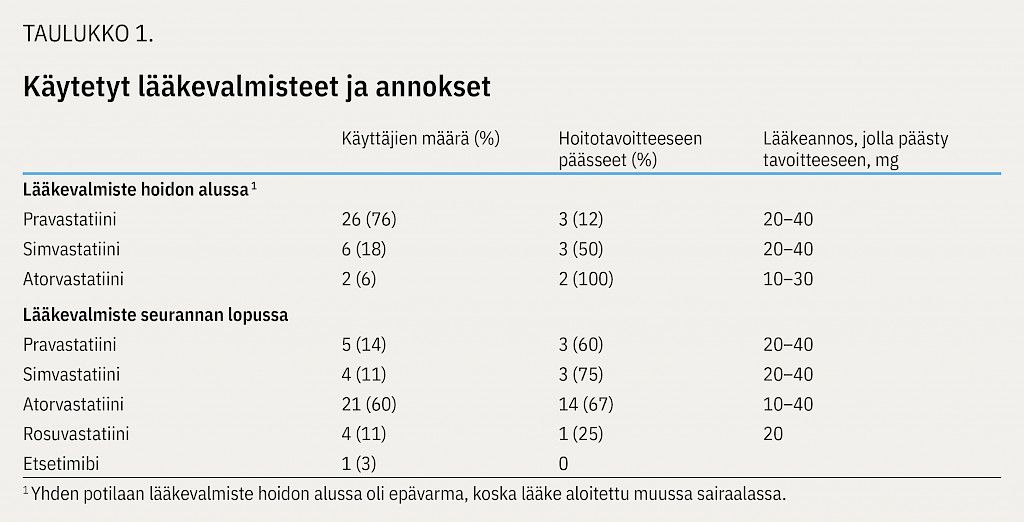
Ensimmäiseksi lääkkeeksi aloitettiin seurantajakson alkupuolella usein simvastatiini, myöhemmin pravastatiini (taulukko 1). Jos tavoitteeseen ei päästy, vaihdettiin atorvastatiiniin ja tarvittaessa rosuvastatiiniin. Etsetimibi aloitettiin vain yhdelle potilaalle, jolle statiini ei sopinut.
Kahdella lääkehoitoa saaneella lapsella (6 %) esiintyi lihaskipuja, joilla saattoi olla yhteys hoitoon. Vatsakipuja esiintyi kahdella potilaalla. Kuudella potilaalla (17 %) todettiin ALAT-arvon nousu kaksinkertaiseksi ja yli rajan 40 U/l, mutta nousua ei pidetty kenelläkään kliinisesti merkittävänä ja lääkitystä voitiin jatkaa. Yhdellä potilaalla todettiin liikunnan jälkeen plasman kreatiniinikinaasitason nousu kaksinkertaiseksi ja yli rajan 400 U/l. Vain yhdellä potilaalla statiinilääkitys vaihdettiin etsetimibiin lihaskivun ja tälle altistavan todetun geenipolymorfian vuoksi. Muilla haitat olivat ohimeneviä.
Kun FH-diagnoosin vuosina 2000–2020 saaneiden lapsipotilaiden määrä (45) suhteutetaan sairaanhoitopiirissä seuranta-aikana syntyneiden lasten määrään (noin 2 100/v), arviolta 64 % tapauksista löydettiin lapsuusiässä, kun sairauden oletettu ilmaantuvuus on 1:600 vastasyntynyttä (13).
Pohdinta
FH:n lääkehoidon aloittaminen lapselle perustuu lääkärin, lapsen ja vanhempien yhteiseen päätökseen. Statiinihoito on lapsuusiässä tehokasta ja turvallista sekä tämän selvityksen että aiempien tutkimusten perusteella (11).
Pravastatiini on ollut ensisijainen statiinivalmiste lasten FH:n hoidossa (3,10). Tulostemme perusteella sillä ei kuitenkaan useimmiten päästä hoitotavoitteeseen, ja myös tehokkaampien statiinien, kuten atorvastatiinin ja rosuvastatiinin, käytöstä lapsilla on viime vuosina julkaistu tutkimuksia (11). Tehokkaampien statiinien valinta vaikuttaa perustellulta jo lääkehoitoa aloitettaessa.
Vaikka aineiston kaikki potilaat eivät päässeet hoitotavoitteeseen seurannan aikana, oli lääkehoidon aikainen LDL-kolesterolin vähenemä hyvä verrattuna vuonna 2019 tehtyyn meta-analyysiin, jossa yhdeksän tutkimuksen keskimääräinen LDL-kolesterolitason lasku oli 32 % (11).
Suomessa on arvioitu, että noin joka 600. henkilö kantaa sairauden aiheuttavaa FH-geenivirhettä (13), tavallisimmin LDL-reseptorigeenissä. Kuitenkin tilastojen perusteella vain puolella heistä on FH-diagnoosi (3,14). Tutkimuksessamme arvioimme lapsuusiässä löydetyn noin 64 % tapauksista. Jotta myös loput tunnistettaisiin, tulisi joko tehostaa vanhempien diagnostiikkaa ja heille tarjottavaa perinnöllisyysneuvontaa tai harkita väestötasoista FH-taudin seulontaa jo lapsuusiässä.
Yleisestä lasten ja nuorten dyslipidemiaseulonnasta on jonkin verran näyttöä, mutta sen kustannustehokkuudesta ja tarkkuudesta ei ole vielä riittävästi tietoa, eikä seulontaa siksi ole pidetty aiheellisena (3,15,16). Perusterveydenhuollon tehtävänä on FH-sukujen seulonta, mutta diagnoosin varmistaminen, hoidon järjestäminen ja seuranta lapsuus- ja nuoruusiässä tapahtuu erikoissairaanhoidossa.
Tämän retrospektiivisen tutkimuksen tulosten luotettavuuteen vaikutti aineiston pieni koko sekä lääke- ja elintapahoidon toteutumisen epävarmuus. Vahvuutena voidaan pitää pitkää seuranta-aikaa verrattuna kontrolloituihin lääketutkimuksiin.
Geneettisissä tutkimuksissa noin neljäsosalla potilaista (27 %) löytyi muu kuin LDLR-geenin valtamutaatio. Näiden mutaatioiden esiintymisen kuvaaminen Suomen väestössä sekä diagnosoimattomien FH-sukujen etsinnän järjestäminen toimivasti vaativat jatkoselvityksiä.
Lapsuusiässä FH-taudin lääkehoidon aloittaneiden potilaiden pitkäaikaisseuranta on myös tärkeää. Tähän haasteita tuo potilaiden seurannan siirtyminen yleensä perusterveydenhuoltoon nuoruusiän jälkeen. Aikuisten FH-potilaiden hoidon keskittämisestä näyttää olevan hyötyä (17). Se voi parantaa taudin kohdistettua seulontaa siten, että yhä useampi FH-potilas saadaan hoitoon jo lapsuusiässä.
Joona Lyly, Jarmo Jääskeläinen: Ei sidonnaisuuksia.
Tanja Kuiri-Hänninen: Matka-, majoitus- ja kokouskulut (Eli Lilly, Merck, Novo Nordisk, Pfizer).
Tämä tiedettiin
• Kolesterolin alentaminen FH-potilailla ehkäisee ateroskleroosia.
• FH-lapsilla LDL-kolesterolitaso nousee jo syntymän jälkeen.
• Statiineista pravastatiinin käytön turvallisuudesta lapsilla on pitkäaikaisinta näyttöä.
Tutkimus opetti
• FH-taudin kohdennettua seulontaa lapsilla tulisi tehostaa.
• Statiinihoidolla päästään tyydyttävään hoitotasapainoon suurimmalla osalla lapsipotilaista, mutta pravastatiinin teho on usein riittämätön.
- 1
- Juonala M, Viikari J, Simell O ym. Lapsuuden elintavat vaikuttavat valtimotaudin kehittymiseen. Suom Lääkäril 2012;67:1485–9.
- 2
- Neil A, Cooper J, Betteridge J ym. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J 2008;29(21):2625–33.
- 3
- Suomalaisen Lääkäriseuran Duodecimin ja Suomen Sisätautilääkärien Yhdistys ry:n asettama työryhmä. Dyslipidemiat. Käypä hoito- suositus 14.12.2022. www.kaypahoito.fi
- 4
- Braamskamp MJAM, Langslet G, McCrindle BW ym. Effect of rosuvastatin on carotid intima-media thickness in children with heterozygous familial hypercholesterolemia: The CHARON Study (Hypercholesterolemia in children and adolescents taking rosuvastatin open label). Circulation 2017;136:359–66.
- 5
- Gidding SS, Champagne MA, de Ferranti SD ym. The agenda for familial hypercholesterolemia: A scientific statement from the American Heart Association. Circulation 2015;132(22):2167–92.
- 6
- Vuorio AF, Gylling H, Turtola H ym. Stanol ester margarine alone and with simvastatin lowers serum cholesterol in families with familial hypercholesterolemia caused by the FH-North Karelia mutation. Arterioscler Thromb Vasc Biol 2000;20(2):500–6.
- 7
- Gylling H, Plat J, Turley S ym. European Atherosclerosis Society Consensus Panel on phytosterols. Plant sterols and plant stanols in the management of dyslipidaemia and prevention of cardiovascular disease. Atherosclerosis 2014;232(2):346–60.
- 8
- Rodenburg J, Vissers MN, Wiegman Ay ym. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation 2007;116(6):664–8.
- 9
- Lozano P, Henrikson NB, Dunn J ym. Lipid screening in childhood and adolescence for detection of familial hypercholesterolemia: A systematic evidence review for the U.S. preventive services task force [Internet]. Rockville (MD): Agency for healthcare research and quality (US) 2016 Report no.: 14-05204-EF-2.
- 10
- Luirink IK, Wiegman A, Kusters DM ym. 20-year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med 2019;381(16):1547–56.
- 11
- Vuorio A, Kuoppala J, Kovanen PT ym. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev 2019;11:CD006401.
- 12
- Dunkel L, Sankilampi U, Saari A. Milloin lapsi on ylipainoinen tai lihava? Duodecim 2010;126:739–41.
- 13
- Lahtinen AM, Havulinna AS, Jula A ym. Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 2015;238(1):64–9.
- 14
- Nordestgaard BG, Chapman MJ, Humphries SE ym. Familial hypercholesterolemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Consensus statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–90.
- 15
- Ritchie SK, Murphy EC, Ice C ym. Universal versus targeted blood cholesterol screening among youth: The CARDIAC project. Pediatrics 2010;126(2):260–5.
- 16
- Wiegman A, Gidding SS, Watts GF ym. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J 2015;36(36):2425–37.
- 17
- Kolehmainen L, Rautiainen P. Familiaalisen hyperkolesterolemian hoitotulokset. Vuosien 2015 ja 2005 vertailu Pohjois-Karjalan keskussairaalan lipidipoliklinikassa. Suom Lääkäril 2017;20:1296–9.
Treatment target achieved in most children with familial hypercholesterolaemia
Background Untreated familial hypercholesterolaemia (FH) leads to atherosclerosis, which may be prevented by lifestyle modifications and medical treatment. Information on FH treatment and its efficacy in Finnish children is scarce.
Methods We collected FH diagnoses in paediatric patients in KUH from 2000 to 2020. Family data, gene mutations, plasma lipid values, and medication were analysed.
Results Of the 45 patients whose FH diagnosis was confirmed, 21 (47%) were girls. Mean age at diagnosis was 7.5 years (range 1.9 to 15.1 years). Of the Finnish major LDL receptor gene mutations, FH-North Karelia was the most common (52%), followed by FH-Helsinki (14%), and FH-Pogosta (7%). Every fourth mutation was not one of the Finnish major mutations. Pravastatin was chosen as the first drug in 76% of the children, but the most common drug during transition to the care of an adult unit was atorvastatin (60%). In medically treated patients, mean plasma LDL-cholesterol was 5.4 mmol/l at the beginning of the follow-up, and 3.0 mmol/l at the end (decrease 43%).
Conclusions Gene testing for common mutations cannot detect all children with FH. The treatment target is achieved in most patients. The position of pravastatin as a primary medication in children should be critically considered
Joona Lyly, Tanja Kuiri-Hänninen, Jarmo Jääskeläinen
Joona Lyly
Medical Student
University of Eastern Finland





