ALS-potilaan monimuotoiset oireet
•Amyotrofiseen lateraaliskleroosiin (ALS) liittyy monia erityisongelmia, jotka vaativat moniammatillista arviointia ja hoitoa.
•Samanaikaista kognitiivista heikentymistä esiintyy useammin kuin aiemmin on ymmärretty.
•Yleisin taustalla oleva geenivirhe, C9ORF72-geenin toistojaksomutaatio, löytyi vuonna 2011.
•Vaikka taudin kulkuun vaikuttavia lääkityksiä ei rilutsolia lukuun ottamatta ole, potilaan moniin oireisiin voidaan antaa helpotusta.
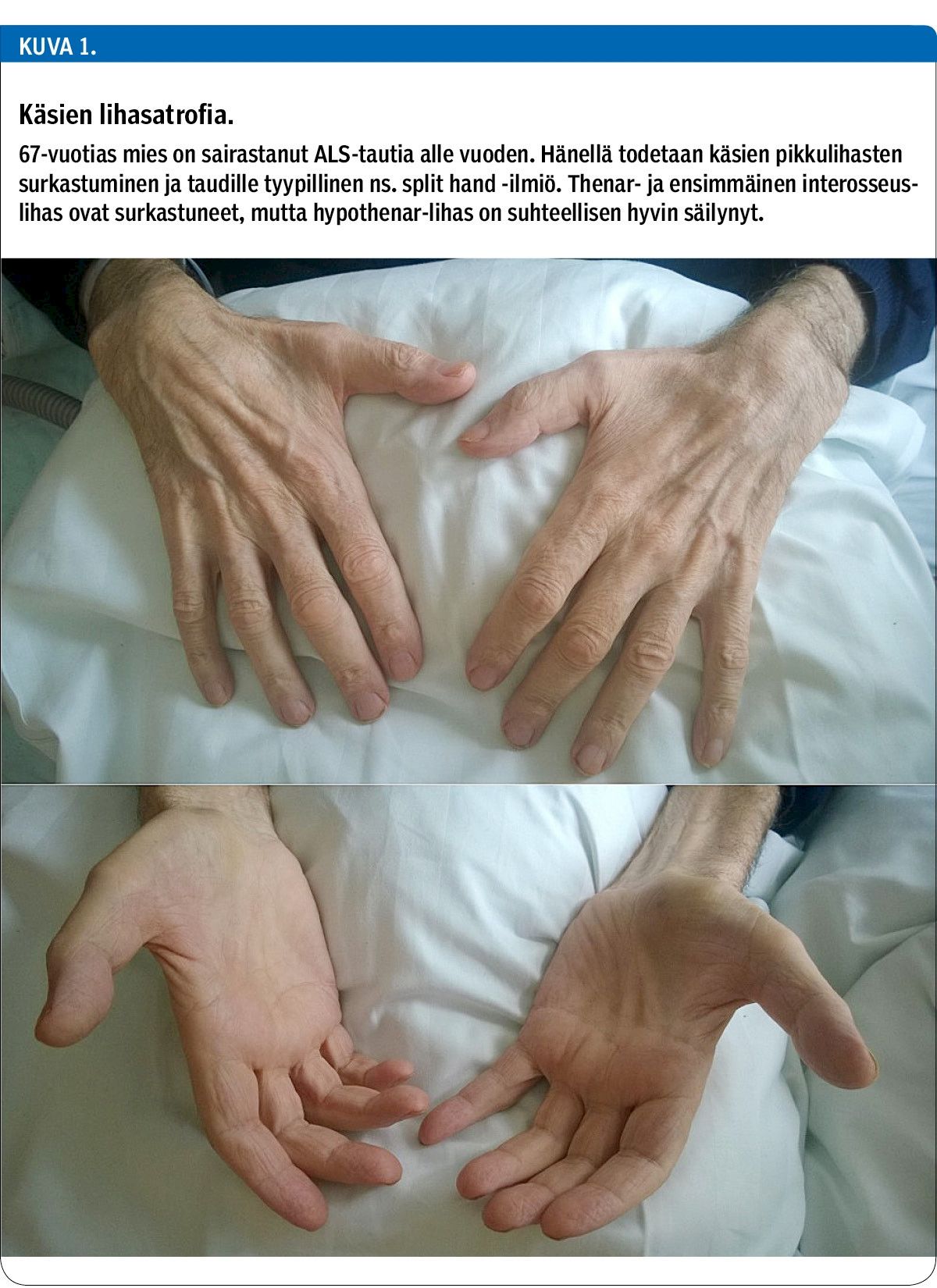 67-vuotias mies on sairastanut ALS-tautia alle vuoden. Hänellä todetaan käsien pikkulihasten surkastuminen ja taudille tyypillinen ns. split hand -ilmiö. Thenar- ja ensimmäinen interosseus-lihas ovat surkastuneet, mutta hypothenar-lihas on suhteellisen hyvin säilynyt.
67-vuotias mies on sairastanut ALS-tautia alle vuoden. Hänellä todetaan käsien pikkulihasten surkastuminen ja taudille tyypillinen ns. split hand -ilmiö. Thenar- ja ensimmäinen interosseus-lihas ovat surkastuneet, mutta hypothenar-lihas on suhteellisen hyvin säilynyt.
Amyotrofinen lateraaliskleroosi (ALS) on yleisin motoneuronitaudeista. Se on harvinainen aikuisiällä alkava sairaus, jossa liikehermot degeneroituvat aiheuttaen raajasta tai bulbaarialueelta alkavan lihasheikkouden ja surkastuman. Tyypillinen tauti etenee vääjäämättä ja melko nopeasti kaikkiin tahdonalaisiin lihaksiin, myös hengityslihaksiin, ja keskimääräinen elinikä oireiden alkamisesta on 2–5 vuotta. ALS:sta tunnetaan kuitenkin myös epätyypillisiä muotoja, joissa eteneminen on hitaampaa, ja noin 10 % tautiin sairastuneista elää yli 10 vuotta, joskin ainakin puolet heistä vaikeavammaisina (1).
ALS-taudin etiologia on edelleen tuntematon, mutta tautia aiheuttavia geenimutaatioita löydetään yhä enemmän, ja niiden avulla pyritään selvittämään taudin syntymekanismeja. Tutkimus on edennyt viime vuosina huimasti, erityisesti sen jälkeen kun taudin taustalla oleva yleisin geenivirhe, kromosomissa 9 sijaitsevan C9ORF72-geenin toistojaksomutaatio, löytyi muutama vuosi sitten (2,3). Mutaation patofysiologisia mekanismeja ja yleisyyttä tutkitaan useissa tutkimusryhmissä. Esimerkiksi yhden selitysmallin mukaan soluissa syntyy viallista RNA:ta, joka sitoutuu tumassa ADARB2-proteiiniin, entsyymiin joka säätelee liikehermojen glutamaattireseptorien toimintaa ja aktiivisuutta. Mutaation vuoksi proteiinin toiminta häiriintyy aiheuttaen glutamaattireseptorien yliaktiivisuutta ja neuroneissa syntyy eksitatorinen vaurio (4).
Suomessa ALS-tautiin sairastuu vajaat 200 henkilöä vuosittain. Maailmanlaajuisesti vuosittainen ilmaantuvuus on 1–3/100 000 ja esiintyvyys 3–8/100 000 henkilöä (5). Tauti todetaan keskimäärin 65 vuoden iässä. Sen itsenäiset riskitekijät ovat ikä, miessukupuoli ja noin 10 %:lla potilaista suvussa esiintyvä tauti (6).
ALS on aiemmin jaettu sporadiseen (SALS) ja familiaaliseen (FALS) tautiin sen mukaan, onko kyseessä yksittäinen tapaus vai useampia samassa suvussa. Tämä jako ei ole nykyään riittävän tarkka, koska tauti voi olla perinnöllinen, vaikka potilas olisi sukunsa ainoa sairastunut (7). Lähiomaisen sairastumisriski vaihtelee suuresti sen mukaan, onko taustalla geenivirhe vai ei. Geneettisen muuntelun osuudeksi on arvioitu vähintään 21 % (8,9).
ALS-tautiin liittyy liikunta- ja toimintakyvyn heikkenemisen lisäksi monia erityisongelmia, jotka erityisesti taudin loppuvaiheessa vaativat asiantuntevaa moniammatillista arviointia ja hoitoa.
Kliiniset oireet ja tutkiminen
ALS-potilaan kliinisessä tutkimuksessa löydöksiä on vaihtelevasti ylemmän ja alemman motoneuronin vauriosta, koska koko liikeradasto motoriselta aivokuorelta selkäytimen etusarven soluihin vaurioituu. Motoneuronit degeneroituvat, korvautuvat glioosilla ja aiheuttavat raajasta tai bulbaarialueelta alkavan lihasheikkouden. Noin 70 %:lla potilaista oireet alkavat yksittäisen raajan (spinaalinen alku), usein ylä- tai alaraajan distaaliosan heikkoudella ja lihasten surkastumisella (kuva 1). Lopuilla potilailla ensioireet tulevat bulbaarialueelta puheen kangistumisena sekä nielun ja kielen toiminnan heikkenemisenä (10).
Tyypillisessä edenneessä taudissa on huomattava lihasatrofia, lihasheikkous ja faskikulaatiot. Edennyt alemman motoneuronin vaurio voi peittää ylemmän merkit alleen, eikä esimerkiksi Babinskin merkkiä saada esiin. Toisaalta pelkät faskikulaatiot eivät ole merkki ALS-taudista. Statuksessa voi olla epäsuhtaa esimerkiksi siten, että jo selvästi surkastuneessa raajassa jänneheijasteet voivat olla hyvinkin vilkkaat. Lopulta kaikki luurankolihakset heikentyvät ja surkastuvat, myös hengityslihakset ja nielun lihaksisto. Kipu tai sensoriset oireet eivät kuulu taudin piirteisiin, mutta kipua voi ilmaantua esimerkiksi spastisuuden, lihaskramppien ja heikentyvän liikkumisen seurauksena.
Diagnoosi on kliininen ja sen tärkeimpänä tukena käytetään ENMG-tutkimusta, joka osoittaa yleensä hyvin alemman motoneuron laaja-alaisen vaurion. ALS-taudille on luotu viralliset kriteerit (11), joita voidaan käyttää diagnostiikan apuna. Tautia epäiltäessä potilas kuuluu lähettää erikoissairaanhoidon jatkotutkimuksiin ja hoitoon.
Genetiikka ja perinnöllisyysneuvonta
Vuonna 1993 löytyi ensimmäinen ALS-geeni, kromosomissa 21 sijaitseva Cu/Zn-superoksididismutaasientsyymiä (SOD1) koodaava geeni, ja pitkään sitä pidettiin ainoana taudin patogeneesiin vaikuttavana tekijänä. Nykyään ALS-tautia pidetään geneettisesti heterogeenisenä, ja noin 50 geenin on esitetty joko aiheuttavan sitä tai muokkaavan sen ilmiasua (12).
Suomessa on löytynyt toistaiseksi kolmen geenin, SOD1, C9ORF72 ja FUS, mutaatioita. Osa ALS-geeneistä, kuten C9ORF72, aiheuttaa myös otsa-ohimolohkodementiaa (frontotemporaalidementia). Tätä mutaatiota esiintyy erityisen paljon Suomessa ja muissa eurooppalaisperäisissä väestöissä. Suomessa vallitsevasti periytyvä C9ORF72-mutaatio löytyy 46 %:lta familiaalista ALS-tautia sairastavista potilaista ja viidesosalta sporadista muotoa sairastavista (13).
Homotsygoottinen SOD1 D91A -mutaatio (D90A) aiheuttaa Suomessa väistyvästi periytyvää ALS-tautia, vaikka SOD1-geenin mutaatiot tyypillisemmin aiheuttavat vallitsevasti periytyvää tautia. Tavallisesti familiaalinen ja sporadinen ALS eivät eroa kliiniseltä oireistoltaan toisistaan, mutta SOD1 D91A:n aiheuttama tauti on poikkeus. Sille on tyypillistä hitaasti etenevä, aina alaraajoista alkava heikkous, jossa alkuun ylemmän motoneuronin vaurion piirteet ovat korostuneena. Suomalaisten SOD1 D91A -mutaation aiheuttamaa tautia sairastavien potilaiden on todettu elävän sairastumisen jälkeen keskimäärin 13 vuotta, ja useat ovat eläneet jopa 20 vuotta (14).
ALS-tautia epäiltäessä geenitutkimukset eivät ole ongelmattomia. Diagnostiikkaan riittää tyypillinen kliininen oireisto täydennettynä ENMG-tutkimuksella eikä geenitutkimuksia välttämättä tarvita (10). Tilanne muuttuu, jos suvussa on tiedossa muita ALS-taudin tai frontotemporaalidementian oirein sairastuneita tai tauti käyttäytyy epätyypillisesti ja diagnostiikkaa halutaan varmentaa geenitutkimuksella.
C9ORF72-mutaation testaaminen on perusteltua vallitsevasti periytyvää tautia ja frontotemporaalidementiaa epäiltäessä. Tulkintavaikeuksia aiheuttavat epätäydellinen penetranssi ja se, että monistuman normaalivaihtelun aluetta ei vielä hyvin tunneta (9,15). Ei myöskään tiedetä, kenelle C9ORF72-mutaatiota kantavalle kehittyy ALS tai frontotemporaalidementia, ja siksi terveiden perheenjäsenten seulontaa ei toistaiseksi suositella.
On erittäin tärkeää etukäteen huomioida positiivisen tuloksen seuraukset muille perheenjäsenille (16). Sporadista tautia sairastavien ja heidän perheidensä geenitestauksista ei ole yhteneväistä suositusta (15,16). SOD1 D91A -mutaation testaaminen on perusteltua, koska sen löytyminen homotsygoottisena ennustaa hidasta etenemistä ja sitä, että lapset ovat kantajia eivätkä heterotsygootteina sairastu.
Perinnöllisyysneuvonta on tärkeää positiivisen geenituloksen jälkeen, mutta se on suositeltavaa myös epäselvissä tilanteissa jo etukäteen, kuten pohdittaessa mahdollisia ennakoivia testauksia. Tulevaisuudessa geenitaustan selvittäminen voi kuitenkin tulla merkittäväksi yksilöllisten, räätälöityjen hoitomuotojen valinnassa.
Kognitiiviset oireet
Aiemmin on ollut vallalla käsitys, että ALS-tautiin ei juuri liity kognitiivisia oireita tai dementiatasoista häiriötä. Viime vuosina käsitys on muuttunut ja tietoa kognitiivisista häiriöistä on tullut runsaasti (17,18).
Monet muutkin ALS-tautiin liitetyt geenit kuin C9ORF72 voivat aiheuttaa myös frontotemporaalidementiaa, mikä osoittaa neuropatologian yhteneväiseksi (12). Tutkimusten mukaan 10–15 %:lla ALS-potilaista todetaan frontotemporaalidementian diagnostiset kriteerit täyttävä oirekuva (17,18). Tämän lisäksi jopa puolella potilaista voidaan todeta frontotemporaalialueen oireita ilman dementiatasoista häiriötä. Tyypillisiä ovat eksekutiivisten toimintojen heikkeneminen, persoonallisuuden ja käyttäytymisen muutokset ja myös kielelliset vaikeudet (17,18). Nämä oireet jäävät helposti huomaamatta fyysisen toimintakyvyn nopean heikentymisen takaa.
ALS-potilaan kohtaamisessa, hoitovalinnoissa ja erityisesti loppuvaiheen hoitoa suunniteltaessa kognitiivisten oireiden mahdollisuus on tärkeää huomioida. Dementoituvaa ALS-potilasta ei pidä ohjata pysyvään hengityskonehoitoon, koska tällöin menetetään mahdollisuus asialliseen keskusteluun hoidon lopettamisesta. Neuropsykologista tutkimusta suositellaan tarvittaessa, erityisesti tilanteissa, joissa tarvitaan kannanottoa esimerkiksi edunvalvontaan tai ajokykyyn.
Frontotemporaalidementiaa ja ALS-tautia sairastavan potilaan ennuste on huonompi kuin pelkkää ALS-tautia sairastavan (19).
Taudin kulkuun vaikuttava lääkehoito
ALS-taudin kulkuun vaikuttavia lääkevalmisteita on etsitty pitkään, mutta toistaiseksi ainoastaan rilutsoli on osoittanut vähäistä tehoa taudin hidastamisessa. Se sai myyntiluvan EU:ssa vuonna 1996, kun kahdessa monikeskustutkimuksessa oli osoitettu lääkettä saavien potilaiden elinikä muutamia kuukausia pidemmäksi kuin lumeryhmässä (20).
Näyttöön perustuvan arvion mukaan rilutsolia suositetaan käytettäväksi ALS-potilaiden hoidossa (A-tason näyttö) (10). Se on turvallinen ja yleensä hyvin siedetty lääke. Sivuvaikutuksina on todettu maksa-arvojen lievää nousua, väsymystä ja heikkoutta sekä vatsaoireita (10,21). Jos rilutsolin käytön aikana ilmenee poikkeuksellista lihasheikkoutta, lääkitys on syytä lopettaa.
Eläinkokeissa rilutsolin on todettu suojaavan rotan motoneuroneja liiallisen glutamaattivälitteisen eksitaation aiheuttamalta solukuolemalta. Uusien geenilöytöjen myötä toiveet taudin patofysiologian ymmärtämisestä ja sitä myötä hoidollisten vaihtoehtojen löytymisestä ovat kiihtyneet ja tutkimustyö tällä saralla on kiivasta (22). Ajatusmalli yhdestä kaikille sopivasta lääkehoidosta on siirtynyt yksilöllisempään geeniperimän mukaiseen hoitoon tai useammalla mekanismilla vaikuttavaan multiterapiaan (4).
Oireenmukainen hoito
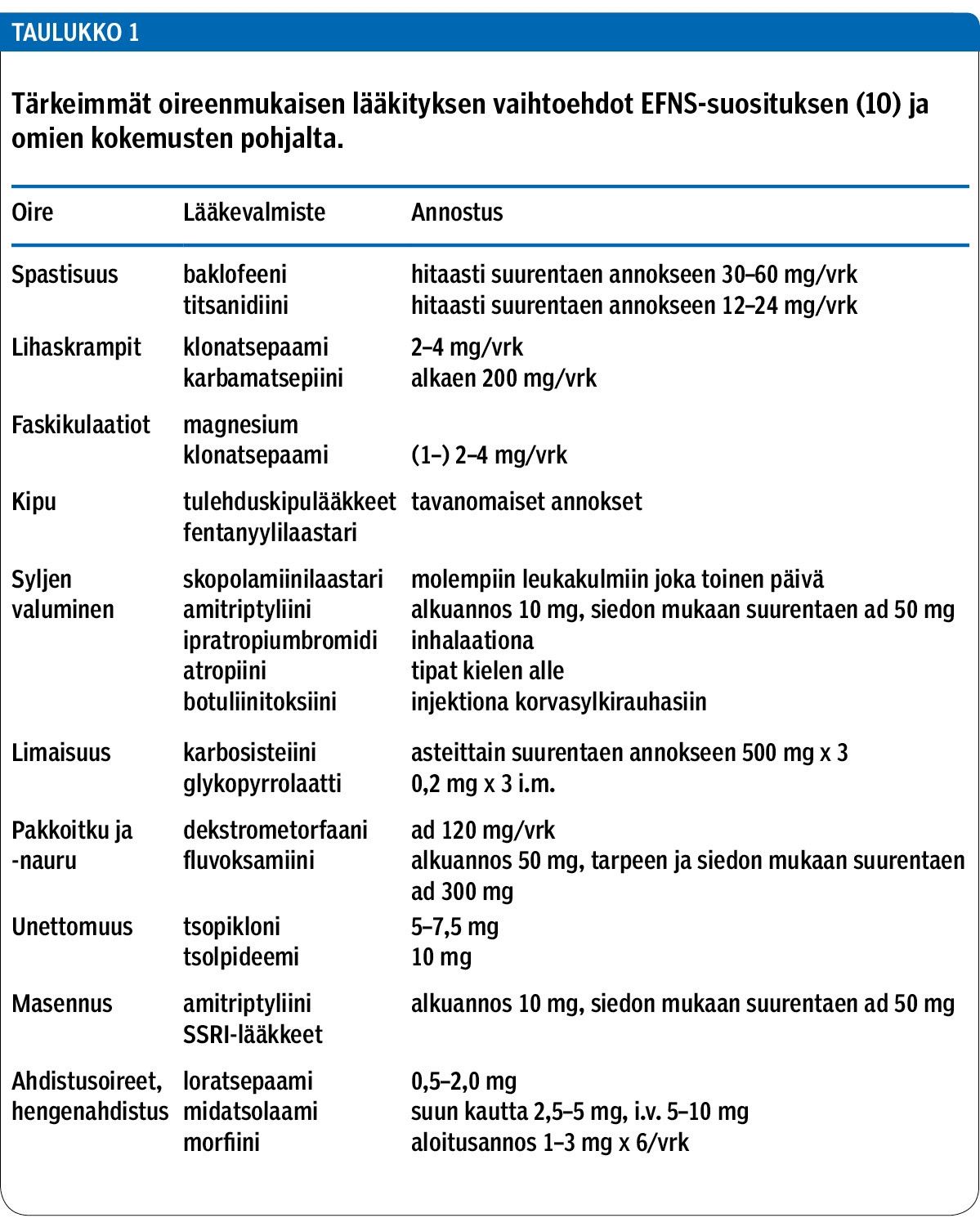
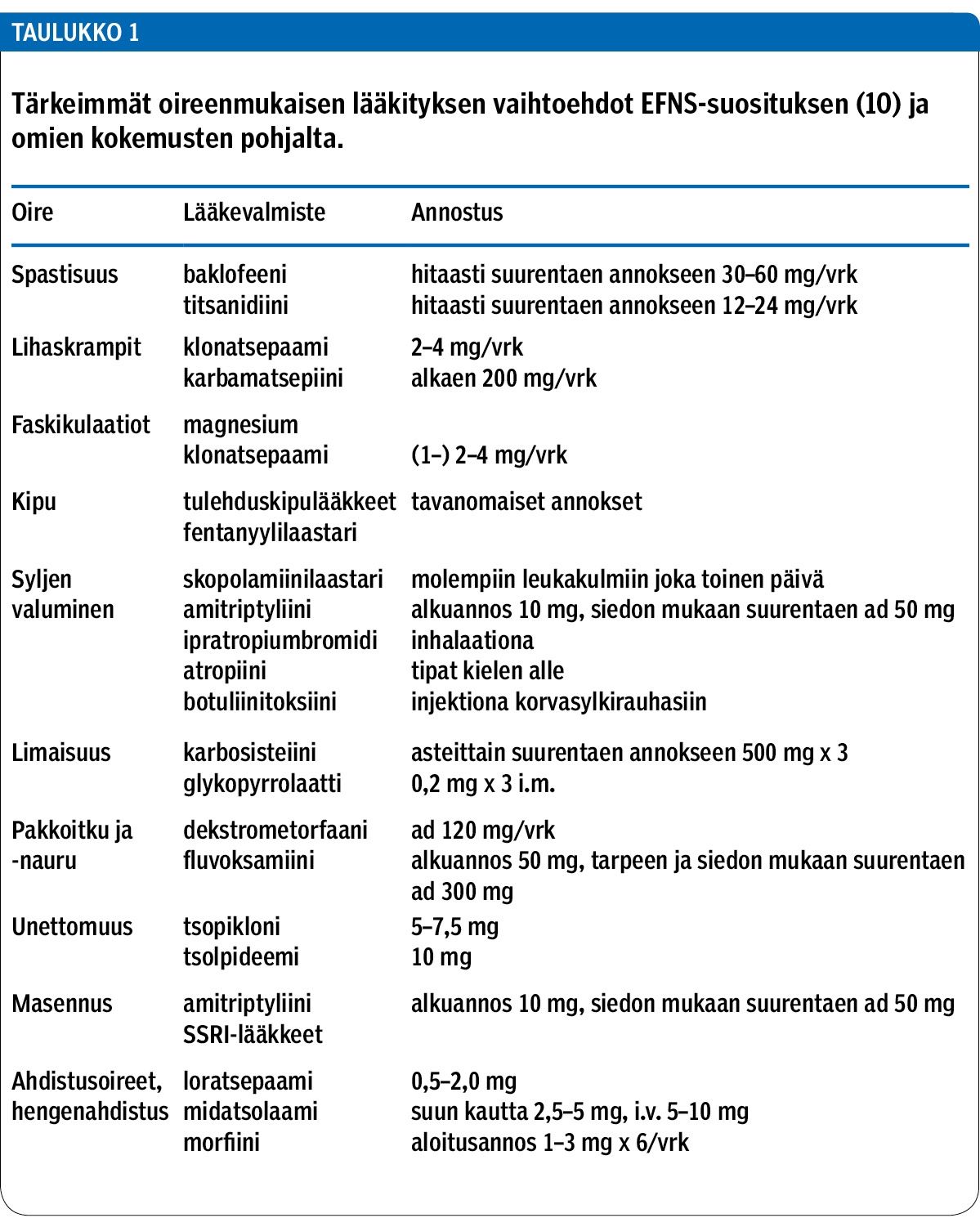
Lihasheikkouteen ei ole toistaiseksi olemassa hoitoa, mutta muita oireita voidaan helpottaa (taulukko 1). Yleensä ALS-potilaan hoitoon ja seurantaan osallistuu moniammatillinen työryhmä: lääkäri, sairaanhoitaja, fysioterapeutti, toiminta- ja puheterapeutti, ravitsemussuunnittelija, sosiaalityöntekijä ja kuntoutusohjaaja. Seuraavassa esitetään taudin monimuotoisiin oireisiin hyväksi havaittuja hoito-ohjeita, jotka ovat hyvin yhteneväisiä eurooppalaisten hoitosuositusten kanssa (10).
Liikunta- ja toimintakyvyn heikentyminen
Joillakin potilailla raajojen spastisuus ja kramppitaipumus aiheuttavat kipua. Spastisuutta voidaan lievittää titsanidiinilla tai baklofeenilla, vaihtoehtona voi käyttää myös diatsepaamia tai muita bentsodiatsepiiniryhmän valmisteita. Lihaskramppeihin voi käyttää esimerkiksi klonatsepaamia tai karbamatsepiinia, myös levetirasetaamia voi kokeilla (10). Faskikulaatiot, (lihasnykinät, elohiiret) kuuluvat ALS-tautiin, mutta ne ovat harvoin potilasta häiritseviä. Tarvittaessa niihin voi käyttää magnesiumia. Muuttunut liikkuminen ja toimintakyvyn heikkeneminen voivat aiheuttaa muskuloskeletaalista kipua, joihin annetaan tavanomaista kivun hoitoa.
Fysioterapialla ja suorituskyvyn mukaan tehtävillä omaehtoisilla harjoitteilla pyritään ehkäisemään virheasentojen ja kipujen syntymistä, vähennetään spastisuutta ja pyritään tukemaan toimintakykyä mahdollisimman pitkään. Kodinmuutostyöt ja apuvälineet sekä liikkumiseen että päivittäisten toimintojen helpottamiseen ovat tarpeen. Liikkumattomuuden myötä laskimotukosten riski kasvaa, mutta profylaktisen lääkityksen hyödyllisyydestä ei ole näyttöä. Riski kannattaa kuitenkin huomioida esimerkiksi käyttämällä tukisukkia.
Kommunikoinnin vaikeutuminen
Bulbaarialkuisessa tautimuodossa puhe vaikeutuu jo hyvin varhaisessa vaiheessa. Puhe muuttuu epäselväksi, dysartriseksi, puheääni hiljenee ja muuttuu monotonisemmaksi. Ongelma on siis puheen tuotossa, ei kielen ymmärtämisessä. Puhekyky voi kadota jo siinä vaiheessa, kun raajoissa ei ole vielä merkittävässä määrin oireita. Myös raajasta alkavassa tautimuodossa puheen vaikeutuminen tulee vääjäämättä oirekuvaan mukaan (23).
Kommunikointikyvyn ylläpitäminen loppuun asti on keskeistä potilaan autonomian kannalta. Ongelmaan on syytä varautua jo ennen ymmärrettävän puheen täydellistä katoamista. Puheterapeutti seuraa puhe- ja nielufunktion muutoksia, antaa opastusta puheen ylläpitämiseksi mahdollisimman pitkään ja siirryttäessä käyttämään kommunikaatioapuvälineitä. Vaihtoehtoja on nykyaikaisista, esimerkiksi tablettiin saatavista sovelluksista yksinkertaisiin kuva- tai kirjaintauluihin (24). Myös läheisten opettaminen näiden kommunikaatiomenetelmien käyttöön on tärkeää.
Nielun toiminta ja pseudobulbaarioireet
Jo taudin alkuvaiheessa potilaan paino saattaa pudota jopa 15–20 kg, todennäköisesti lihasmassan kuihtumisen, nielemisvaikeuksien aiheuttaman ravitsemusvajeen ja muuttuneen energiametabolian vuoksi (25). Taudin seurannassa ravitsemuksen turvaaminen on keskeistä. Puheterapeutti neuvoo nielemistekniikoista ja ruoan ja nesteiden koostumuksesta. Erityisesti nesteiden sakeuttajien käyttö on hyödyllistä. Ravitsemuslisiä voidaan käyttää ja riittävästä nesteytyksestä huolehditaan.
Myös aspiraatioriskin säännöllinen arvioiminen on tarpeen. Perkutaaninen endoskooppinen gastrostomia eli PEG-katetrin asennus on käytännössä tarpeen siinä vaiheessa, kun ravitsemus ei suun kautta ole enää turvallista tai riittävää. Usein PEG-katetrin asennus siirtyy psykologisista syistä, mutta asennus on hankalampaa, kun tauti etenee pidemmälle, erityisesti jos mukana on jo merkittävää hengitysvajausta.
Syljen valuminen suun alueen lihasheikkouden tai spastisuuden vuoksi on monelle ikävä ja sosiaalisesti hankala haitta. Sitä voidaan vähentää käyttämällä suuta kuivattavia valmisteita, tavallisimmin amitriptyliinia tai skopolamiinilaastaria. Myös sylkirauhasten botuliini-injektioita voidaan käyttää, mutta sivuvaikutuksena voi olla nielemisvaikeuden lisääntyminen. Sylkirauhasten sädetys on yksi vaihtoehto.
Limaisuus on usein bulbaari- ja hengitysoireisilla lisääntynyt ja yskimisvoima heikentynyt. Limaa irrottavaa karbosisteiinilääkitystä voi kokeilla, mutta limaisuutta voidaan vähentää myös asentotyhjennyshoidoilla ja imua käyttäen.
Ns. pseudobulbaarioireisiin liittyy kiusallista pakkoitkua tai -naurua ja haukottelua, jota voidaan vähentää trisyklisillä masennuslääkkeillä, SSRI-lääkkeillä tai dekstrometorfaania sisältävällä yskänlääkkeellä. Omaisille on hyvä kertoa, että oireet eivät ole merkki kognitiivisen tason laskusta tai henkisistä ongelmista.
Psyykkiset oireet
Mielialaongelmat, ahdistus, unettomuus ja uupumusoireet ovat vakavaa tautia sairastavalla tyypillisiä, ja ne on myös tärkeää huomioida hoidossa. Ennusteen kertominen ja loppuvaiheen suunnittelu hyvissä ajoin sekä mahdollisuus etukäteen varautua toimintakyvyn, syömisen, kommunikoinnin ja hengityksen muutoksiin voivat lievittää ahdistusta ja antaa potilaalle mahdollisuuden osallistua hoitopäätösten tekemiseen. Ensitietopäivät ja sopeutumisvalmennuskurssit antavat tietoa tulevista muutoksista ja tukitoimista. Anksiolyytit, unilääkkeet ja terminaalivaiheessa morfiini ovat käytetyimpiä lääkehoitoja.
Hengitysvajaus
ALS-potilaan tavallisin kuolemansyy on hengityslihaksiston heikkenemisestä aiheutuva hengityksen loppuminen. Bulbaarialkuisen ALS-taudin ennuste on huonompi kuin spinaalisen ja hengitysvaikeudet ilmaantuvat nopeammin. Hengityslihaksista alkavan lihasheikkouden ennuste on kaikkein huonoin. Etenevä toiminta- ja liikuntakyvyn heikkeneminen johtaa liikkumisen vähenemiseen, joka puolestaan vähentää tarvittavaa hengitysreserviä. Näin ollen potilas ei välttämättä tunne hengenahdistusta kuin vasta taudin myöhäisessä vaiheessa.
Hengitysvajauksen kehittymisen säännöllinen seuranta ja tarvittaessa sen palliatiivinen hoito on suositeltavaa aloittaa jo varhaisessa vaiheessa (10,26). Hoidon tarkoituksena on ensisijaisesti helpottaa hengenahdistusta ja parantaa elämänlaatua. Potilaan kanssa tulee keskustella jo hyvissä ajoin ennen oireiden ilmaantumista loppuvaiheen hoidosta hengityksen heiketessä. Potilaan toiveet kirjataan ja hoitolinjausta tarkistetaan säännöllisesti.
Kajoamaton ventilaatiohoito on suositeltava hoitomuoto, jos koneellista hengityksen tukihoitoa annetaan. Sen on todettu parantavan elämänlaatua, mutta myös lisäävän elinaikaa 200–800 vuorokautta (27). Suomalaisessa artikkelissa ja pääkirjoituksessa on hiljattain käsitelty yksityiskohtaisesti ALS-potilaan hengitysvajauksen hoitoa, hoidon kriteereitä ja linjauksia (26,28).
Loppuvaiheen hoito
Useimmiten elämän loppuvaiheen hoito tapahtuu terveyskeskuksen vuodeosastolla. Osa potilaista hakeutuu saattokoteihin, osa toivoo loppuvaiheen hoitoa kotona. Hoitopaikasta on syytä keskustella hyvissä ajoin. Hengenahdistustunne lisääntyy hiilidioksidipitoisuuden kasvaessa ja samalla usein ahdistus ja kuolemanpelkokin lisääntyvät.
Hengenahdistuksen hoidoksi voidaan ensin käyttää loratsepaamia tai midatsolaamia ja loppuvaiheessa tavallisimmin morfiinia. Opiaateista riittävät yleensä pienet annokset, jolloin ei tarvitse pelätä lääkkeen aiheuttamaa hengityslamaa. Hiilidioksidiretention aiheuttamaan levottomuuteen ja sekavuuteen voi käyttää neuroleptejä. Kipuihin voidaan käyttää esimerkiksi fentanyylilaastaria. Tehokas loppuvaiheen limaisuutta vähentävä lääke on glykopyrrolaatti.
Lopuksi
ALS-tauti etenee täydelliseen liikunta- ja kommunikaatiokyvyttömyyteen. Tautia epäiltäessä suositellaan pikaista neurologin konsultaatiota diagnoosin varmistamiseksi ja hoitolinjojen laatimiseksi. Potilaan ja taudin tuntevan lääkärin on syytä kertoa diagnoosi. Sen jälkeen suositellaan moniammatillista erikoissairaanhoidossa tapahtuvaa seurantaa ja hoitoa, johon sisältyy myös tarvittava geneettinen informaatio ja perinnöllisyysneuvonnan tarjoaminen.
Vaikka taudin kulkuun vaikuttavia lääkityksiä ei rilutsolia lukuun ottamatta ole, voidaan potilaan moniin oireisiin antaa helpotusta. Rilutsolihoidon aloittamisesta tulisi keskustella mahdollisimman aikaisin, ja hoito pitää lopettaa viimeistään vuodepotilaalta, jolla suojeltavia motoneuroneja ei enää merkittävästi ole.
Ennakoimalla tulevia ongelmia potilaalle annetaan mahdollisuus sopeutua ja vaikuttaa hoitovalintoihin. Jo melko alussa on syytä keskustella potilaan toiveesta, minkälaista hengitystukea hän haluaa. On tärkeää huomioida isolle osalle potilaista oirekuvaan liittyvä kognitiivisen tason heikkeneminen. Henkisen tuen antaminen, potilaan toiveiden kunnioittaminen ja autonomian säilyminen on oleellista. Myös omaisten jaksamisen tukeminen on tärkeä osa palliatiivista hoitoa.
Johanna Palmio: Apurahat (Maire Taponen säätiö), luentopalkkiot (Orion Pharma, Genzyme), matka-, majoitus- ja kokouskulut (Genzyme).
Hannu Laaksovirta: Ei sidonnaisuuksia.
- 1
- Couratier P, Corcia P, Lautrette G, Nicol M, Preux PM, Marin B. Epidemiology of amyotrophic lateral sclerosis: A review of literature. Rev neurol (Paris) 2016;172:37–45.
- 2
- Renton AE, Majounie E, Waite A ym. A Hexanucleotide Repeat Expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–68.
- 3
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF ym. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–56.
- 4
- Tienari P, Kiviharju A, Valori M, Lindholm D, Laaksovirta H. Tumapatologia ja glutamaattiherkkyys. Duodecim 2016;132:423–31.
- 5
- Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci 2001;191:3–9.
- 6
- Armon C. An evidence-based medicine approach to the evaluation of the role of exogenous risk factors in sporadic amyotrophic lateral sclerosis. Neuroepidemiology 2003;22:217–28.
- 7
- Byrne S, Elamin M, Bede P, Hardiman O. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2012;83:365–7.
- 8
- Keller MF, Ferrucci L, Singleton AB ym. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol 2014;71:1123–34.
- 9
- Al-Chalabi A, Fang F, Hanby MF ym. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry 2010;81:1324–6.
- 10
- The EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis. EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS) – revised report of an EFNS task force. Eur J Neurol 2012;19:360–75.
- 11
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2000;1:293–9.
- 12
- Boylan K. Familial amyotrophic lateral sclerosis. Neurol Clin 2015;4:807–30.
- 13
- Laaksovirta H, Peuralinna T, Schymick JC ym. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol 2010;9:978–85.
- 14
- Laaksovirta H. Perheittäin esiintyvä ja periytyvä amyotrofinen lateraaliskleroosi (FALS ja SOD1-ALS). Suom Lääkäril 2007;62:2555–8.
- 15
- Chiò A, Battistini S, Calvo A ym. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 2014;85:478–85.
- 16
- Rohrer JD, Isaacs AM, Mizielinska S ym. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 2015;14:291–301.
- 17
- Phukan J, Elamin M, Bede P ym. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012;83:102–8.
- 18
- Goldstein LG, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol 2013;12:368–80.
- 19
- Elamin M, Phukan J, Bede P ym. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 2011;76:1263–9.
- 20
- Laaksovirta H. Rilutsoli – ensimmäinen ALS-lääke. Suom Lääkäril 1998;53:2979.
- 21
- Bensimon G, Lacomblez L, Meininger V, the ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–91.
- 22
- Riva N, Agosta F, Lunetta C, Filippi M, Quattrini A. Recent advances in amyotrophic lateral sclerosis. J Neurol 2016;263:1241–54.
- 23
- Makkonen T, Korpijaakko-Huuhka AM, Ruottinen H ym. Oral motor functions, speech and communication before a definitive diagnosis of amyotrophic lateral sclerosis. J Commun Disord 2016;61:97–105.
- 24
- Luotonen M, Aitola L. Puhe puuttuu, motoriikka mättää – kommunikoinnin apuvälineet vahvistavat elämänhallintaa. Duodecim 2013;129:169–75
- 25
- Vercruysse P, Sinniger J, El Oussini H ym. Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain 2016;139:1106–22.
- 26
- Siirala W, Korpela J, Vuori A ym. Amyotrofinen lateraaliskleroosi ja hengitysvajaus. Duodecim 2015;131:127–35.
- 27
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 2006;5:140–7.
- 28
- Laaksovirta H, Kainu A. Etenevän hengityshalvauksen hoidon linjaus. Duodecim 2015;131:115–6.
Diverse manifestations in ALS patients
Amyotrophic lateral sclerosis (ALS) is a rare motor neuron disease that causes progressive muscle weakness and atrophy. Eventually the disorder affects all skeletal muscles and results in various manifestations including decreased respiratory functions, dysarthria and dysphagia. Therefore, it is recommended that after the diagnosis the patient is followed-up and treated by a multidisciplinary care team. Riluzole is the only drug available for prolonging survival in ALS. However, almost all symptoms of the disease can be alleviated. Decline in cognitive functions is seen more often than previously understood. Approximately 50% of patients with ALS have some cognitive impairment, and 10–15% evince criteria for diagnosing frontotemporal dementia which is important to take into consideration when planning and discussing treatment options with the patient. Maintaining the patient’s autonomy and ability to communicate is important. Mutations in SOD1 and repeat expansions in C9ORF72 are found in Finnish ALS patients. Of the two C9ORF72 is more prevalent and accounts for 46% of familial and 21% of sporadic cases.





{kind=link}