Rakenteellisten synnynnäisten sydänvikojen genetiikka
• Synnynnäiset sydänviat ovat vastasyntyneiden yleisimpiä rakennepoikkeamia. Ne voivat esiintyä yksittäin ilman liitännäisvikoja tai osana oireyhtymää.
• Yksittäin esiintyvien sydänvikojen periytyminen on monitekijäistä. Niiden syntyyn vaikuttavat sekä geenit että ympäristötekijät.
• Molekyylikaryotyypitys tehdään, mikäli sydänvikaa sairastavalla todetaan muitakin rakenteellisia poikkeamia ja sydänvian epäillään olevan osa oireyhtymää.
• Yksittäin esiintyvissä sydänvioissa geenidiagnostiikka ei ole vielä laajamittaisessa käytössä.

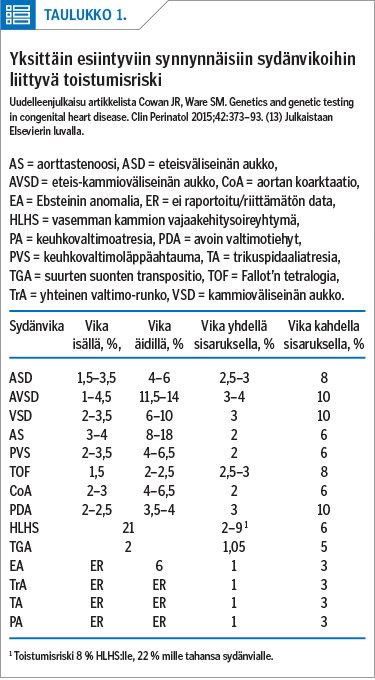
TAULUKKO 1.
Yksittäin esiintyviin synnynnäisiin sydänvikoihin liittyvä toistumisriski
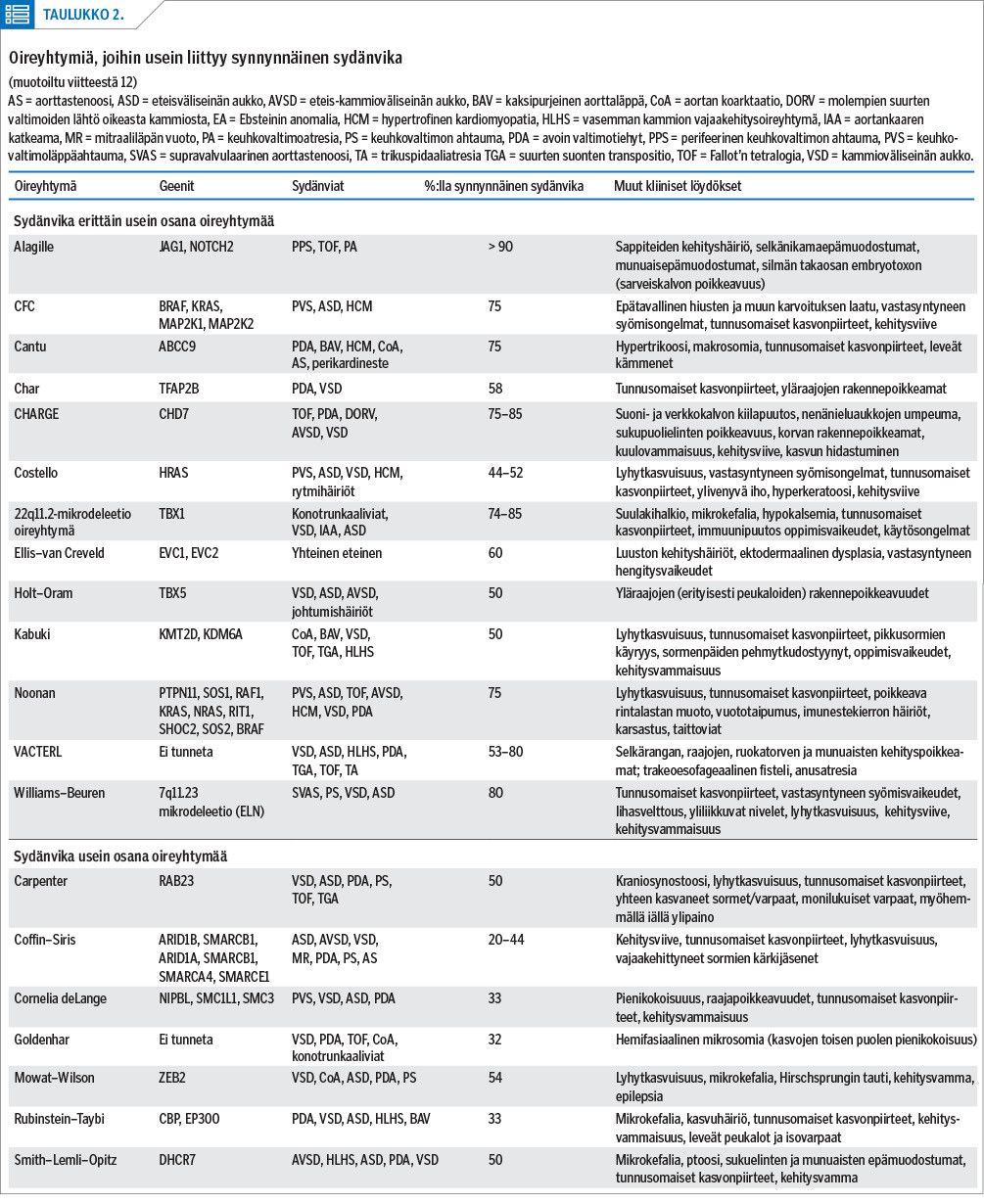
TAULUKKO 2.
Oireyhtymiä, joihin usein liittyy synnynnäinen sydänvika
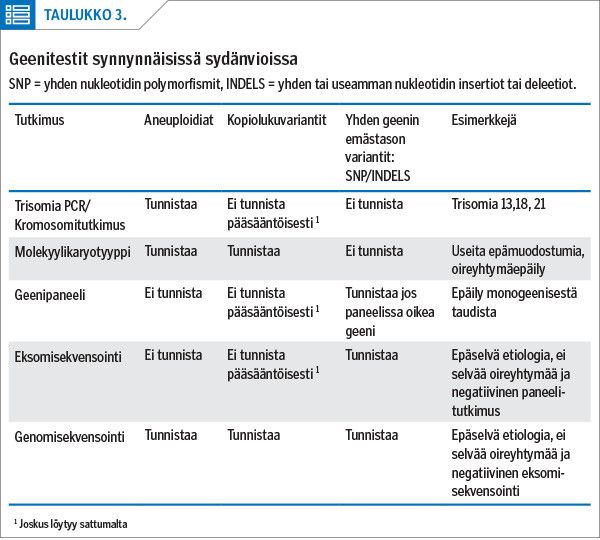
TAULUKKO 3.
Geenitestit synnynnäisissä sydänvioissa

Synnynnäisiä sydänvikoja esiintyy arviolta 1 %:lla vastasyntyneistä; ne ovat vastasyntyneiden yleisimpiä rakennepoikkeamia (1). Luku ei pidä sisällään kaksipurjeista aorttaläppää, joka on yleensä lapsuudessa oireeton ja siksi alidiagnosoitu. Sen esiintyvyyden väestötasolla arvellaan olevan noin yhden prosentin luokkaa (2,3).
Sydämen rakenteet kehittyvät ensimmäisen raskauskolmanneksen aikana. Sydämen kehitys on monivaiheinen tapahtumaketju, johon vaikuttavat useiden eri solujen ja molekulaaristen signalointireittien välisen vuorovaikutuksen lisäksi sydämen läpi virtaavan veren hemodynaamiset voimat (4,5). Normaalia kehitystä voivat häiritä ulkoiset tekijät, kuten teratogeenit tai äidin sairaudet, ja geenien tai kromosomien poikkeavuudet. Ympäristötekijät, kuten äidin poikkeava raskaudenaikainen glukoosiaineenvaihdunta, diabetes, korkea ikä ja lihavuus sekä eräät raskauden aikana käytetyt lääkkeet, foolihapon puute tai sairastetut infektiot lisäävät synnynnäisten sydänvikojen riskiä (6,7,8,9,10).
Noin neljännes vastasyntyneisyyskauden aikana diagnosoiduista sydänvioista vaatii leikkaushoidon ensimmäisen elinvuoden aikana. Lääketieteellisten hoitojen ja sydänkirurgian kehittymisen ansiosta suurin osa vaikeaakin sydänvikaa sairastavista lapsista selviytyy aikuisikään. Merkittävimpiin synnynnäisiin sydänvikoihin liittyy kuitenkin huomattavaa sairastavuutta ja elinajan odote on lyhentynyt.
Synnynnäiset sydänviat ovat monitekijäisesti periytyviä rakennevikoja
Sydämen rakenneviat voivat liittyä oireyhtymiin (20 %) tai esiintyä yksittäin, ilman muita rakennevikoja (80 %) (11). Yksittäin esiintyvät sydämen rakenneviat ovat etiologialtaan heterogeenisiä, eli ne voivat olla seurausta monesta erilaisesta tekijästä. Koska sydämen rakenneviat esiintyvät usein osana oireyhtymää, liitännäisvikojen mahdollisuus on tärkeä arvioida.
Oireyhtymiin liittyvien sydänvikojen taustalla voi olla minkä tahansa tyyppinen geneettinen syy: kromosomien lukumäärän poikkeavuus eli aneuploidia, kromosomin osien pieni häviämä tai monistuma eli kopiolukumuutos tai yhden geenin patogeeninen variantti (12). Sen sijaan yksittäin esiintyvien sydämen rakennevikojen taustalla kopiolukumuutosten ja yhden geenin emäsvarianttien merkitys korostuu (13).
Epidemiologisten tutkimusten mukaan synnynnäiselle sydänvialle löydetään geneettinen syy 20–30 %:ssa tapauksista (13). Potilaista 10–15 %:lla on aneuploidia, kuten Downin oireyhtymä, Turnerin oireyhtymä, 13- tai 18-trisomia (14). Patologisia kopiolukumuutoksia löytyy 3–25 %:lta tutkittavista, joilla synnynnäinen sydänvika esiintyy osana oireyhtymää, ja 3–10 %:lta niistä, joilla on synnynnäinen sydänvika ilman liitännäisvikoja (15). Yhden geenin aiheuttamia vikoja löytyy arviolta 3–10 %:lta potilaista (16,17).
Synnynnäinen sydänvika periytyy todennäköisemmin äidiltä kuin isältä
Puhtaasti mendelistisesti periytyviä yksittäin esiintyviä sydänvikoja tavataan harvoin. Useimmiten näissä tapauksissa taustalla on todennäköisesti useampi vialle altistava geenivariantti (oligogeeninen tauti) tai vialle altistava geenivariantti yhdessä ympäristötekijän tai jonkin muun epigeneettisen tekijän kanssa johtaa sydänvian kehittymiseen (18,19,20,21).
Mikäli toisella vanhemmista on synnynnäinen sydänvika, periytymisriski jälkeläiselle on 5–10 %. Sydänviat periytyvät kuitenkin useammin äidiltä kuin isältä (taulukko 1). Mitä useammalla perheenjäsenellä todetaan sydänvika, sitä suurempi on toistumisriski.
Suvuittain esiintyville rakennevioille on tyypillistä heikentyneen läpäisevyyden eli penetranssin lisäksi ilmiasun vaihtelevuus. Samaan geneettiseen varianttiin voi liittyä erilainen sydänvika eri perheissä, mutta myös perheiden sisällä (19). Tanskalaistutkimuksen mukaan perheittäin esiintyvistä sydänvioista vain 50 %:ssa fenotyyppi on konkordantti eli samanlainen (22).
Periytymisriski vaihtelee huomattavasti myös sydänvian tyypin mukaan. Suurin periytymisriski on heterotaksiaan eli elinten poikkeavaan sijaintiin liittyvissä sydänvioissa, oikean ulosvirtauskanavan rakennevioissa, vasemman ulosvirtauskanavan ahtauman tyyppisissä vioissa (LVOTO) ja konotrunkaali- eli valtasuonten ulosvirtausalueen vioissa (23).
Perimän muutokset oireyhtymiin liittyvissä sydämen rakennevioissa
Aneuploidiat
Kromosomien lukumäärän poikkeavuudet eli aneuploidiat ovat vanhin tunnettu synnynnäisten sydänvikojen geneettinen syy. Koska aneuploidiat vaikuttavat suureen määrään geenejä, lähes kaikilla niihin liittyy sydänvikojen lisäksi myös muita anomalioita (24).
Yleisin aneuploidia on 21-trisomia eli Downin oireyhtymä. Jopa noin puolella 21-trisomiapotilaista on synnynnäinen sydänvika (25), yleisimmin oikovirtausvika: eteis-kammioväliseinän aukko (AVSD), kammioväliseinän aukko (VSD), eteisväliseinän aukko (ASD) tai avoin valtimotiehyt (PDA). Myös Fallot’n tetralogian (TOF) riski on lisääntynyt näillä potilailla. Oireiltaan vaikeampiin 13- ja 18-trisomioihin liittyy 80–90 %:lla sydänvikoja (26,27).
Turnerin oireyhtymässä (monosomia X) noin kolmanneksella potilaista todetaan synnynnäinen sydänvika, yleisimmin vasemman ulosvirtauskanavan ahtauman tyyppinen (LVOTO), kuten kaksipurjeinen aorttaläppä (BAV), aortan koarktaatio (CoA) ja harvemmin vasemman kammion vajaakehitysoireyhtymä (HLHS) tai osittain poikkeava keuhkolaskimopaluu (PAPVD). On tärkeää muistaa, että arviolta 3–8 %:lle Turner-potilaista kehittyy seurannassa nousevan aortan laajentuma (aneurysma). Se on mahdollinen, vaikka Turner-potilaalla ei olisi kaksipurjeista aorttaläppää, joten elinikäinen seuranta on aiheellinen.
Kopiolukumuutokset
Mikrodeleetiot ja -duplikaatiot ovat kromosomien osien pienikokoisia rakenteellisia muutoksia, häviämiä tai kahdentumia, jotka ovat yleensä yli 1 000 nukleotidia pitkiä. Kopiolukumuutokset voivat olla periytyviä tai uusia (de novo).
Oireyhtymien taustalta on tunnistettu useita hyvin kuvattuja kopiolukumuutoksia. Yleisin on kromosomin 22q11.2 mikrodeleetio. Sen esiintyvyys on noin 1 : 6 000 elävänä syntynyttä lasta (28), ja se löytyy arviolta 2 %:lta kaikista sydänvikapotilaista (29), mutta jopa 13–56 %:lta konotrunkaalivikapotilaista (30). Tämän mikrodeleetion kliininen ilmentymä vastaa DiGeorgen oireyhtymää ja valokardiofasiaalista oireyhtymää, mutta fenotyyppi voi vaihdella paljonkin jopa saman perheen sisällä (31). DiGeorge-fenotyypin potilaista 10 %:lta ei kuitenkaan löydy 22q11.2-mikrodeleetiota. 22q11.2-mikrodeleetioon liittyvistä sydänvioista 70 % on konotrunkaalivikoja, kuten Fallot’n tetralogia, yhteinen valtimorunko (truncus arteriosus), subaortaalinen kammioväliseinän aukko, aortankaaren B-tyypin katkeama (IAA) ja muut aortankaaren anomaliat.
Muita tunnettuja sydänvikoja aiheuttavia kopiolukumuutoksia ovat mm. Williams–Beuren oireyhtymä (7q11.23-mikrodeleetio), johon liittyy supravalvulaarinen aorttastenoosi (SVAS) ja pulmonaalistenoosi (PS), ja Jacobsenin oireyhtymä (11q:n terminaalinen deleetio), johon liittyy tyypillisimmin kammioväliseinän aukko, vasemman ulosvirtauskanavan ahtauman tyyppinen vika tai kolmasosalla muunlainen synnynnäinen sydänvika (32).
Yleisesti tavattujen kopiolukumuutosten lisäksi sydänvikapotilailla on harvinaisia de novo -kopiolukumuutoksia useammin kuin terveillä (33,34,35). Tutkimusten perusteella näiden varianttien arvioidaan aiheuttavan ainakin 10–15 % kaikista sydänvioista (34,35).
Yhden geenin muutokset
Kopiolukumuutosten lisäksi yhden geenin muutokset, pistemutaatiot ja yhden tai useamman nukleotidin insertiot tai deleetiot aiheuttavat oireyhtymiä, joihin liittyy synnynnäisiä sydänvikoja.
Rasopatiat ovat autosomissa vallitsevasti periytyviä oireyhtymiä, esimerkiksi Noonanin oireyhtymä, kardiofasiokutaaninen oireyhtymä (CFC) ja Costellon oireyhtymä (CS) (36). Rasopatia-nimitys tulee siitä, että näihin sairauksiin liittyvät geenit kuuluvat RAS-signalointipolkuun, jonka geenit säätelevät ihmisen kasvua ja kehitystä.
Rasopatioiden ilmiasu vaihtelee ja niihin liittyvät ongelmat ovat laaja-alaisia: sydänvika, tyypilliset kasvonpiirteet, syömishäiriöt, kasvuhäiriö, endokrinologiset ongelmat, hematologiset poikkeavuudet, munuaisongelmat, ortopediset ongelmat sekä neurokognitiiviset poikkeavuudet (36). Oireyhtymään liittyvät sydänviat ovat tyypillisimmin keuhkovaltimoläpän ahtauma (PVS) ja hypertrofinen kardiomyopatia (HCM), harvemmin aortan koarktaatio, eteisväliseinän aukko, Fallot’n tetralogia ja mitraaliläpän rakennepoikkeavuudet (12). Costello-oireyhtymää voivat vielä komplisoida eteisperäiset rytmihäiriöt.
Rasopatioiden diagnostiikkaan on tarjolla geenipaneeleja, jotka sisältävät tunnetut oireyhtymään liittyvät geenit.
Heterotaksia
Heterotaksiat ovat sairauksia, joissa elinten sijainti poikkeaa normaalista vasen–oikea-akselilla. Heterotaksiapotilailla on todettu vallitsevasti ja peittyvästi periytyviä yhden geenin variantteja (37,38), aneuploidioita, kromosomien uudelleenjärjestymistä ja kopiolukumuutoksia (39,40).
Vasemmassa isomerismissa rintakehän elimet ovat vasentyyppiset, eli potilaalla on kaksi vasentyyppistä eteiskorvaketta, puuttuva sinussolmuke ja useita pernoja. Oikeassa isomerismissa on kaksi oikeatyyppistä eteistä korvakkeineen, kaksi sinussolmuketta ja perna puuttuu. Isomerismipotilaiden ilmiasussa esiintyy huomattavaa vaihtelua. Molempiin isomerismeihin liittyy sydämen rakennevikoja, noin 80 %:lla potilaista (41). Peilikuvasijainti-tyyppiseen (situs inversus) heterotaksiaan voi liittyä myös Kartagenerin oireyhtymä värekarvatoimintahäiriöineen.
Pierpontin ym. katsauksessa on käsitelty laajasti geneettisiä oireyhtymiä, joihin liittyy synnynnäisiä sydänvikoja (taulukko 2) (12).
Geenit ja niiden säätely yksittäin esiintyvissä sydämen rakennevioissa
Suurin osa sydänvioista (yli 80 %) esiintyy ilman liitännäisvikoja potilailla, joiden lähisuvussa ei ole esiintynyt vastaavia vikoja aiemmin. On ilmeistä, että de novo -variantit aiheuttavat osan synnynnäisistä sydänvioista. Potilas on myös voinut periä molemmilta vanhemmiltaan useampia sydänvialle altistavia geenivariantteja, jotka eivät yksinään riitä aiheuttamaan vikaa, mutta yhdessä johtavat sairauden kehittymiseen. Toisaalta ympäristöön liittyvät riskitekijät saattavat vaikuttaa vian kehittymiseen erityisesti sellaisilla yksilöillä, joilla on geneettisiä riskivariantteja.
Toisinaan tavataan myös perheitä, joissa toisen tai kolmannen asteen sukulaisella, esimerkiksi indeksipotilaan serkulla tai isovanhemmalla, on synnynnäinen sydänvika. Heikentynyt penetranssi liittyy todennäköisesti taudin oligogeenisyyteen ja ympäristöriskitekijöihin.
Esimerkkinä yksittäisen geenimuutoksen yhteydestä sydänvikoihin on transkriptiotekijä NKX2-5, jonka vallitsevan pistemutaation on todettu aiheuttavan suvuittain yksittäin esiintyviä eteisväliseinän aukkoja, mutta myös muita rakennevikoja, kuten Fallot’n tetralogiaa, sekä eteis-kammiokatkosta (42). Transkriptiotekijöiden GATA4, GATA6 ja NR2F2 geenien vallitsevien pistemutaatioiden on todettu aiheuttavan eteisväliseinän aukkoja ja vasemman ulosvirtauskanavan ahtauman tyyppisiä vikoja (43,44,45). Myös sarkomeerigeenien variantteja on raportoitu synnynnäisten sydänvikojen aiheuttajina, kuten MYH6-variantit eteisväliseinän aukon (46) ja MYH7-variantit Ebsteinin anomalian taustalta (47).
Vallitsevasti periytyvien solusignalointigeenin NOTCH1 varianttien ajatellaan aiheuttavan 5–10 % vasemman ulosvirtauskanavan ahtauman tyyppisistä vioista, sitä useammin, mitä vaikeampi vika on kyseessä (19,48). Suomalaisessa 49:n vasemman kammion vajaakehitysoireyhtymäpotilaan kohortissa todennäköisesti patogeeninen NOTCH1 loss of function -variantti löytyi kolmelta potilaalta (49). Muista solusignalointiin liittyvistä geeneistä JAG1-geenin variantteja on löytynyt Fallot’n tetralogiassa ja pulmonaalistenoosipotilailta (50), SMAD6-geenin variantteja vasemman ulosvirtauskanavan ahtauman tyyppisissä vioissa (51) ja ACVR-1- ja CRELD1-variantteja potilailta, joilla on eteis-kammioväliseinän aukko (52,53).
Tuoreiden tutkimustulosten perusteella vaikuttaa siltä, että epigeneettiset tekijät vaikuttavat synnynnäisten sydänvikojen kehittymiseen (54,55,56). Epigeneettisen säätelyn vaikutuksesta geenien toiminnan tehokkuus vaihtelee eri yksilöissä tai solulinjoissa eri aikoina. Tärkeimpiä epigeneettisiä mekanismeja ovat DNA:n metylaatio, kromosomien rakennetta ylläpitävien histonien muokkaus ja sekä ei-koodaavan RNA:n aiheuttama RNA-interferenssi.
Sydämen kehityksen kannalta tärkeiden geenien metylaatiotasoissa (54,55) ja toisaalta ei-koodaavan RNA:n ekspressiossa (57,58) on todettu eroja esimerkiksi Fallot-, aorttastenoosi- ja kammioväliseinän aukkotapauksissa potilaiden ja terveiden verrokkien välillä. Tutkimusten perusteella äidin diabetekseen liittyvä jälkeläisen lisääntynyt sydänvian riski voisi ainakin osittain liittyä diabeteksen aiheuttamiin epigeneettiseen säätelyn poikkeamiin (59).
Geenidiagnostiikka kliinikon työvälineenä
Molekyylikaryotyypitys tehdään, mikäli sydänvikaisella lapsella todetaan muitakin rakenteellisia poikkeamia tai epäillään oireyhtymää (taulukko 3). Se löytää perimässä esiintyviä pieniä deleetioita tai duplikaatioita ja yleensä myös aneuploidiat, mutta niitä epäiltäessä tehdään ensisijaisesti kromosomitutkimus. Käytännössä molekyylikaryotyypitys on syrjäyttänyt fluoresenssi in situ -hybridisaation (FISH) sydänvikojen diagnostiikassa lähes kokonaan.
Trisomia-PCR-tutkimus voidaan tehdä vastasyntyneelle lapselle, kun tarvitaan nopeita päätöksiä esimerkiksi sydänvian leikkauskelpoisuudesta. Sillä voidaan muutaman vuorokauden kuluessa selvittää kromosomien 21, 13, 18, X ja Y lukumäärät, mutta koska se ei paljasta kromosomien rakenteellisia muutoksia, poikkeavan tuloksen jälkeen esimerkiksi translokaatiot suljetaan pois perinteisellä kromosomitutkimuksella.
Mikäli ilmiasun perusteella epäillään tiettyä yhden geenin tautia, voidaan tehdä geenipaneelitutkimus. Rutiininomaiset geenipaneelitutkimukset ovat kuitenkin sydänvikojen geneettisten syiden diagnostiikassa vasta vähäisessä käytössä. Kaikille geenipaneelitutkimukseen ohjattaville tehdään ensin molekyylikaryotyypitys. Mikäli sekä molekyylikaryotyypityksen että geenipaneelitutkimuksen tulos jää negatiiviseksi, perinnöllisyyslääkäri voi selvittää esimerkiksi perheessä toistuvan hyvin vaikean tai fataalin sydämen rakennevian genetiikkaa eksomi- ja tai lähitulevaisuudessa koko genomin sekvensoinnilla. Tällaisissa tapauksissa tutkimustulosta voitaisiin käyttää hyväksi perinnöllisyysneuvonnassa ja alkiodiagnostiikassa.
Sikiödiagnostiikka
Kaikille odottaville äideille tarjotaan mahdollisuus sikiön rakenneultraääniseulontaan toisen raskauskolmanneksen aikana. Mikäli perheessä on aikaisemmin tunnistettu sydänvika, mutta syynä oleva kromosomipoikkeavuus tai geenivirhe ei ole tiedossa, tehdään sydänvian uusiutumisriskin arvio. Sikiön sydänvian riskin ylittäessä 3 % lääkärin tekemä rakennekaikututkimus ja tarvittaessa perinnöllisyysneuvonta ovat aiheelliset ja uusiutumisriskin arvion ollessa 1–2 % harkinnanvaraiset (60).
Sikiön sydänvikaan liittyvien vaikeiden kehityshäiriöiden diagnostiikassa tarvitaan usein nopeita tuloksia. Tutkimuksia varten sikiön DNA:ta voidaan eristää istukka- tai lapsivesinäytteestä, raskauden keston mukaan. Näytteestä voidaan tutkia perheessä aikaisemmin tunnistettu geenivirhe tai tehdä sydänvian arvion yhteydessä molekyylikaryotyypitys tai jossain tilanteissa geenipaneelitutkimus.
Rakennevikojen geenidiagnostiikan kliininen merkitys
Sydänvikojen geneettisen taustan tuntemus ohjaa hoitoa erityisesti silloin, kun lapsen sydänvika on osa oireyhtymää. Se auttaa kliinikkoa saamaan kokonaisvaltaisemman käsityksen potilaan ongelmien laajuudesta tai seurantaa vaativista ongelmakohdista.
Esimerkiksi konotrunkaalinen sydänvika voi olla vastasyntyneellä 22q11.2-mikrodeleetion ainoa ilmiasu, ja siksi kaikilta konotrunkaalivika- ja aortankaaren B-tyypin katkeamapotilailta tutkitaan molekyylikaryotyypitys. 22q11.2-mikrodeleetio-oireyhtymään liittyvät vastasyntyneen hypokalsemia ja immuunipuolustuksen häiriöt tulee huomioida mahdollisessa leikkaus- ja tehohoidossa, ja ne saattavat vaatia myös pitkäaikaisseurantaa. Oireyhtymään liittyvät neuropsykiatriset ongelmat sekä hidastunut kehitys vaativat lastenneurologin tutkimuksia ja seurantaa. Kun erityisvaikeudet havaitaan ja niihin kohdistetaan tukitoimet varhain, kokonaishaitta potilaalle jää todennäköisesti pienemmäksi.
Sairauden geenitaustan tunteminen helpottaa perinnöllisyysneuvontaa, ja vaikeiden, toistumisriskissä olevien sydänvikojen geenidiagnoosi voi vaikuttaa hoitostrategian valintaan. Geenitutkimukset eivät aina anna selkeää vastausta siihen, aiheuttaako geneettinen löydös potilaan sydänvian tai oireyhtymän. Joskus löydöksen merkitys jää epäselväksi (variant of unknown significance, VUS). Perheen on hyvä olla tietoinen tästä mahdollisuudesta ennen testaukseen ryhtymistä. Yhteistyö perinnöllisyyslääkärien kanssa on välttämätöntä ja avain hyvään diagnostiseen lopputulokseen.
Lopuksi
Synnynnäiset sydänviat ovat monitekijäisesti periytyviä tauteja, jotka voivat esiintyä yksittäin tai osana oireyhtymää. Perintötekijät vaikuttavat merkittävästi synnynnäisten sydänvikojen kehittymiseen. Kliinisessä työssä on tärkeää huomioida, että suurin osa yksittäin esiintyvistä synnynnäisistä sydänvioista esiintyy kuitenkin yksilöillä, joiden suvussa ei aikaisemmin ole sydänvikoja todettu. Sen vuoksi sikiön sydänvikoja ei voida seuloa pelkän sukuhistorian perusteella.
Epäiltäessä oireyhtymää sydänvian taustalla geneettiset selvittelyt ovat tärkeitä, jotta voidaan tunnistaa erityistä seurantaa tai hoitoa vaativat sairaudet. Ilman oireyhtymää esiintyvissä sydänvioissa genetiikkaa tunnetaan vielä huonosti eikä rutiiniluonteista geenidiagnostiikkaa voida suositella. Tutkimus alalla kuitenkin etenee huimaa vauhtia. Todennäköisesti käytännöt muuttuvat tulevaisuudessa, kun saadaan uutta tietoa yksittäisinä pidettyjen sydänvikojen geenivarianttien yhteydestä esimerkiksi neurologisiin ongelmiin (61). Myös epigenetiikan syvällisempi ymmärtäminen voi jatkossa tuoda työkaluja synnynnäisten sydänvikojen diagnostiikkaan ja hoitoon.
Kiitokset perinnöllisyyslääkäreille dosentti Sirpa Ala-Mellolle ja dosentti Anna-Kaisa Anttoselle sekä lasten tehohoitolääkäri LT Johanna Hästbäckalle arvokkaista kommenteista käsikirjoitukseen.
Epigeneettisen säätelyn vaikutuksesta geenien toiminnan tehokkuus vaihtelee.
Sikiön sydänvikoja ei voida seuloa pelkän sukuhistorian perusteella.
Emmi Helle, Tiina Ojala: Ei sidonnaisuuksia.
- 1
- Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J Pediatr 2008;153:807–13.
- 2
- Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol 1984;53:849–55.
- 3
- Steinberger J, Moller JH, Berry JM, Sinaiko AR. Echocardiographic diagnosis of heart disease in apparently healthy adolescents. Pediatrics 2000;105(4 Pt 1):815–8.
- 4
- Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res 2015;117:89–98.
- 5
- Ando J, Yamamoto K. Vascular mechanobiology: endothelial cell responses to fluid shear stress. Circ J 2009;73:1983–92.
- 6
- Øyen N, Diaz LJ, Leirgul E ym. Prepregnancy diabetes and offspring risk of congenital heart disease: a nationwide cohort study. Circulation 2016;133:2243–53.
- 7
- Helle EIT, Biegley P, Knowles JW ym. First trimester plasma glucose values in women without diabetes are associated with risk for congenital heart disease in offspring. J Pediatr 2018;195:275–8.
- 8
- Hautala J, Gissler M, Ritvanen A ym. The implementation of a nationwide anomaly screening programme improves prenatal detection of major cardiac defects: an 11-year national population-based cohort study. BJOG 2019;126:864–73.
- 9
- Persson M, Razaz N, Edstedt Bonamy A-K, Villamor E, Cnattingius S. Maternal overweight and obesity and risk of congenital heart defects. J Am Coll Cardiol 2019;73:44–53.
- 10
- Kalisch-Smith JI, Ved N, Sparrow DB. Environmental risk factors for congenital heart disease. Cold Spring Harb Perspect Biol, verkossa ensin 23.9.2019. doi: 10.1101/cshperspect.a037234
- 11
- Blue GM, Kirk EP, Sholler GF, Harvey RP, Winlaw DS. Congenital heart disease: current knowledge about causes and inheritance. Med J Aust 2012;197:155–9.
- 12
- Pierpont ME, Brueckner M, Chung WK ym. Genetic basis for congenital heart disease: revisited: A Scientific Statement From the American Heart Association. Circulation 2018;138:e653–711.
- 13
- Cowan JR, Ware SM. Genetics and genetic testing in congenital heart disease. Clin Perinatol 2015;42:373–93.
- 14
- Hartman RJ, Rasmussen SA, Botto LD ym. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol 2011;32:1147–57.
- 15
- Jin SC, Homsy J, Zaidi S ym. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 2017;49:1593–601.
- 16
- van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJM. The changing epidemiology of congenital heart disease. Nat Rev Cardiol 2011;8:50–60.
- 17
- Zaidi S, Choi M, Wakimoto H ym. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013;498:220–3.
- 18
- Foffa I, Ait Alì L, Panesi P ym. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet 2013 Apr 11;14-44.
- 19
- Kerstjens-Frederikse WS, van de Laar IMBH, Vos YJ ym. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med 2016;18:914–23.
- 20
- Gifford CA, Ranade SS, Samarakoon R ym. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019;364:865–70.
- 21
- Jarrell DK, Lennon ML, Jacot JG. Epigenetics and mechanobiology in heart development and congenital heart disease. Diseases 2019;7:E52. doi: 10.3390/diseases7030052
- 22
- Brodwall K, Greve G, Leirgul E, Tell GS, Vollset SE, Øyen N. Recurrence of congenital heart defects among siblings-a nationwide study. Am J Med Genet A 2017;173:1575–85.
- 23
- Øyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PKA, Melbye M. Recurrence of congenital heart defects in families. Circulation 2009;120:295–301.
- 24
- Wimalasundera RC, Gardiner HM. Congenital heart disease and aneuploidy. Prenat Diagn 2004;24:1116–22.
- 25
- Vis JC, Duffels MGJ, Winter MM ym. Down syndrome: a cardiovascular perspective. J Intellect Disabil Res 2009;53:419–25.
- 26
- Wyllie JP, Wright MJ, Burn J, Hunter S. Natural history of trisomy 13. Arch Dis Child 1994;71:343–5.
- 27
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis 2012;7:81.
- 28
- Botto LD, May K, Fernhoff PM ym. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003;112:101–7.
- 29
- Agergaard P, Olesen C, Østergaard JR. The prevalence of chromosome 22q11. 2 deletions in 2,478 children with cardiovascular malformations. A population-based study. Am J Med Genet A 2012;158A:498–508.
- 30
- Peyvandi S, Lupo PJ, Garbarini J ym. 22q11.2 deletions in patients with conotruncal defects: data from 1,610 consecutive cases. Pediatr Cardiol 2013;34:1687–94.
- 31
- Digilio MC, Angioni A, De Santis M ym. Spectrum of clinical variability in familial deletion 22q11.2: from full manifestation to extremely mild clinical anomalies. Clin Genet 2003;63:308–13.
- 32
- Grossfeld PD, Mattina T, Lai Z ym. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A 2004;129A:51–61.
- 33
- Soemedi R, Wilson IJ, Bentham J ym. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 2012;91:489–501.
- 34
- Glessner JT, Bick AG, Ito K ym. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 2014;115:884–96.
- 35
- Kim DS, Kim JH, Burt AA ym. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J Thorac Cardiovasc Surg 2016;151:1147–51.
- 36
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009;19:230–6.
- 37
- Mohapatra B, Casey B, Li H ym. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet 2009;18:861–71.
- 38
- Kaasinen E, Aittomäki K, Eronen M ym. Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1). Hum Mol Genet 2010;19:2747–53.
- 39
- Fakhro KA, Choi M, Ware SM ym. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci U S A 2011;108:2915–20.
- 40
- Cowan JR, Tariq M, Shaw C ym. Copy number variation as a genetic basis for heterotaxy and heterotaxy-spectrum congenital heart defects. Philos Trans R Soc Lond B Biol Sci 2016 Dec 19;371(1710)
- 41
- Lin AE, Krikov S, Riehle-Colarusso T ym. Laterality defects in the national birth defects prevention study (1998--2007): birth prevalence and descriptive epidemiology. Am J Med Genet A 2014;164:2581–91.
- 42
- Schott JJ, Benson DW, Basson CT ym. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998;281:108–11.
- 43
- Hirayama-Yamada K, Kamisago M ym. Phenotypes withGATA4 orNKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 2005;135:47–52.
- 44
- Kodo K, Nishizawa T, Furutani M ym. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA 2009;106:13933–8.
- 45
- Al Turki S, Manickaraj AK, Mercer CL ym. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet 2014;94:574–85.
- 46
- Ching Y-H, Ghosh TK, Cross SJ ym. Mutation in myosin heavy chain 6 causes atrial septal defect. Nat Genet 2005;37:423–8.
- 47
- Budde BS, Binner P, Waldmüller S ym. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One 2007;2:e1362.
- 48
- Garg V, Muth AN, Ransom JF ym. Mutations in NOTCH1 cause aortic valve disease. Nature 2005;437:270–4.
- 49
- Helle E, Cordova-Palomera A, Ojala T ym. Loss of function, missense, and intronic variants in NOTCH1 confer different risks for left ventricular outflow tract obstructive heart defects in two European cohorts. Genet Epidemiol 2019;43:215–26.
- 50
- Eldadah ZA, Hamosh A, Biery NJ ym. Familial tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet 2001;10:163–9.
- 51
- Tan HL, Glen E, Topf A ym. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat 2012;33:720–7.
- 52
- Robinson SW, Morris CD, Goldmuntz E ym. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet 2003;72:1047–52.
- 53
- Smith KA, Joziasse IC, Chocron S ym. Dominant-negative ALK2 allele associates with congenital heart defects. Circulation 2009;119:3062–9.
- 54
- Sheng W, Qian Y, Wang H ym. DNA methylation status of NKX2-5, GATA4 and HAND1 in patients with tetralogy of fallot. BMC Med Genomics 2013 Nov 1;6:46.
- 55
- Radhakrishna U, Albayrak S, Alpay-Savasan Z ym. Genome-wide DNA methylation analysis and epigenetic variations associated with congenital aortic valve stenosis (AVS). PLoS One 2016;11(5):e0154010.
- 56
- Lyu G, Zhang C, Ling T ym. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genomics 2018 Jun 4;19(1):428.
- 57
- Zhang J, Chang J-J, Xu F ym. MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of Fallot. Can J Cardiol 2013;29:1695–703.
- 58
- Li D, Ji L, Liu L ym. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS One 2014;9(8):e106318.
- 59
- Schulze KV, Bhatt A, Azamian MS ym. Aberrant DNA methylation as a diagnostic biomarker of diabetic embryopathy. Genet Med 2019;21:2453–61.
- 60
- Donofrio MT, Moon-Grady AJ, Hornberger LK ym. Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation 2014;129:2183–242.
- 61
- Blue GM, Ip E, Walker K ym. Genetic burden and associations with adverse neurodevelopment in neonates with congenital heart disease. Am Heart J 2018;201:33–9.
Genetics in congenital heart defects
Genetics in congenital heart defects
Congenital heart defects (CHDs) are the most common congenital malformations, affecting 0.8–1% of the population. CHDs can be isolated or can present as a part of a syndrome. Isolated congenital heart defects have a complex inheritance pattern with variable phenotype and reduced penetrance. According to recent research, isolated CHDs are in many cases likely of oligogenic origin, meaning that more than one predisposing genetic variant is needed for disease development. In addition to genes, environmental factors such as maternal diabetes, obesity, and certain infections during pregnancy are known to increase the risk for CHDs in the offspring. All types of genetic variants from aneuploidies, such as trisomy 21, to single nucleotide polymorphisms have been associated with CHDs. A karyotype or molecular karyotype test is used if a syndromic CHD is suspected. Genetic testing is currently not in routine clinical use in isolated CHDs.